بیماری کاوازاکی که به آن سندرم کاوازاکی هم گفته میشود یک بیماری جدی است که باعث التهاب دیواره عروق کوچک و متوسط در تمامی بدن و ازجمله عروق کرونر قلب که خونرسانی عضله قلب را تامین می کنند می شود.این بیماری به صورت اولیه کودکان و شیرخواران را درگیر می کند و علت اصلی بیماریهای اکتسابی قلب در کودکان استاین بیماریها را در آغاز سندروم )نشانگان( غدد لنفاوی مخاطی – پوستی می نامیدند.
چونکه این بیماری عقدههای لنفاوی و غشاهای مخاطی داخل دهان، بینی و حلق را هم درگیر می کند.
شیوع و همه گیری شناسی
اگر چه در 80% موارد کودکان زیر 5سال را درگیر می کند اما این بیماری در کودکان بزرگتر و نوجوانان هم روی میدهد گر چه بیماری در این سنین شایع نیست. این بیماری کودکان پسر را بیشتر از دختر بچه ها درگیر می کند و بیشترین موارد درگیری این بیماری درفصل زمستان و اوایل بهار روی می دهد این بیماری ارثی نیست بیماری در کودکان ژاپنی شایعتر است، اما در سراسر دنیا دیده میشود.
اسامی دیگر
- بیماری کاوازاکی
- سندرم عقدههای لنفاوی
- سندرم عقدههای لنفاوی مخاطی پوستی
- سندرم کاوازاکی

علت بیماری
علت بیماری کاوازاکی ناشناخته است؛ اما احتمالاً منشأ عفونی دارد. واکنش ازدیاد حساسیتی یا پاسخ ایمنی مختلشده احتمالاً توسط یک عامل عفونی(مانند باکتریها یا ویروسهای خاص)تحریک می شود و در نتیجه روند التهابی خاصی ایجاد میشود که منجر به تخریب عروق خونی در افرادی می شود که از نظر ژنتیکی مستعد هستند.
علایم ناشی از بیماری
بیماری با یک تب با درجه بالا که علتی ناشناخته دارد و برای حداقل 5 روز طول کشیده باشد شروع می شود. کودک مبتلا معمولاً بسیار تحریک پذیر است. التهاب ملتحمه چشم (قرمز شدن چشمها) می تواند همزمان با تب یا به دنبال آن رخ دهد که همراه چرک یا ترشح چشمی نیست.
انواع مختلفی از بثورات پوستی در کودک دیده می شود که مشابه بثورات سرخجه یا تب مخملک، کهیر، ضایعات پوستی گرد و برجسته و … است. بثورات پوستی اکثراً تنه و اندامهای فوقانی و تحتانی را درگیر می کند و می تواند ناحیه پوشک کودک را نیز تغییرات دهانی در بیماران عبارتند از: لبهای ترک خورده با رنگ قرمز رنگ پریده، زبان قرمز رنگ(که معمولاً “زبان توت فرنگی ”نامیده میشود)و قرمز شدن حلق.
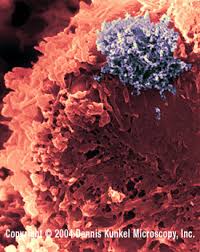
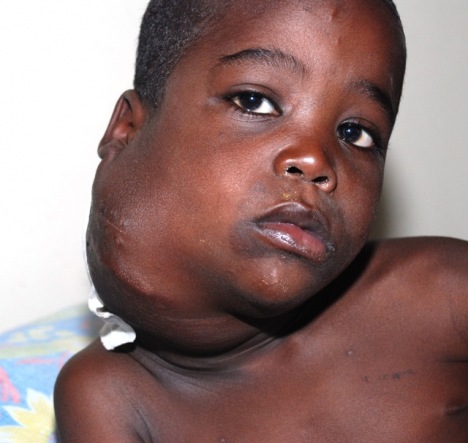
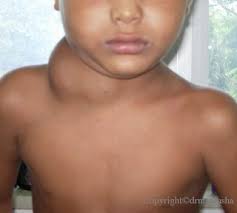
دست ها و پاها نیز ممکن است درگیر شوند که در این صورت کف دست ها و پاها متورم شده، قرمز رنگ میشوند. به دنبال این حالت )در حدود هفته های دوم یا سوم بیماری( ،نوک انگشتان دست و پا دچار پوستهریزی می شود.بیش از نیمی از بیماران دچار بزرگ شدن غدد لنفاوی گردنی می شوند که اندازه هر غده لنفاوی بزرگشده حداقل 5/1 سانتی متر است.گاهی وقتها علایم دیگری مانند درد مفصلی و یا مفاصل متورم، درد شکمی، اسهال، تحری کپذیری و سردرد نیز بروز میکنند .
روش تشخیص
هیچ گونه تست آزمایشگاهی مشخصی برای تشخیص قطعی بیماری وجود ندارد .
تشخیص قطعی بیماری با حضور تب بالای بدون علت شناخته شده که 5 روز یا بیشتر طول کشیده باشد – به همراه حضور
4 مورد از 5 مورد زیر داده میشود:
(1) التهاب دوطرفه ملتحمه چشم ،
(2)غدد لنفاوی بزرگ شده،
(3) بثورات پوستی،
(4)درگیری دهان و زبان ،
و(5)تغییرات اندامهای فوقانی و تحتانی )دست ها و پاها(؛ در صورتی که این موارد را نتوان به بیماری دیگری نسبت داد .
در صورتی که با توجه به تعریف فوق نتوان به تشخیص قطعی دست یافت، باید به انواع ناکامل بیماری شک کرد.
آزمونهای آزمایشگاهی
یافته های آزمایشگاهی برای تشخیص بیماری اختصاصی نیستند، ولی بیانگر وجود درجاتی از التهاب هستند. شاخص های وجود التهاب عبارتند از: افزایش ESR (معمولاً از بیماریهایی با تظاهرات مشابه بالاتر است)، افزایش تعداد گلبولهای سفید خون، و کمخونی(کاهش تعداد گلبولهای قرمز خون) .
تعداد پلاکتهای خون(سلولهایی که در انعقاد خون شرکت می کنند) عموماً در هفته نخست طبیعی است، اما در هفته دوم افزایش قابل توجهی پیدا میکند.
بیماران باید به طور دوره ای تحت معاینه بالینی و ارزیابی آزمایشهای خونی قرار گیرند که این کار تا زمان بهبود آنان باید ادامه یابد. در ابتدای بیماری باید نوار قلب( ECG) و اکوکاردیوگرافی انجام شود.
اکوکاردیوگرافی با نمایش شکل و اندازه شریانهای کرونر قادر به تشخیص آنوریسم می باشد. در موارد ابتلای کودک به ناهنجاری های عروق کرونر، آزمایشها و ارزیابیهای بیشتری مورد نیاز است.
درمان
بیماران مبتلا به بیماری کاوزاکی که تشخیص آنان قطعی یا احتمالی است باید در بیمارستان بستری شوند و برای ارزیابی درگیری احتمالی قلب تحت نظر و مراقبت قرار گیرند.
برای کاهش عوارض قلبی، درمان باید به محض تشخیص آغاز شود. درمان بیماری مشتمل بر آسپیرین و گاماگلوبولین داخل وریدی است که هر دو باید با مقادیر بالا تجویز شوند. هر دو داروی به کار رفته در درمان، التهاب سیستمیک را کاهش میدهند و علایم حاد بیماری را از بین می برند. از آنجا که گاماگلوبولین قادر به جلوگیری از ناهنجاریهای عروق کرونر در درصد بالایی از بیماران است، تجویز آن با مقادیر زیاد یکی از بخشهای اساسی درمان است. استفاده از کورتیکوستروئیدها نیز با رواج کمتر کاربرد دارد.
در تعداد زیادی از بیماران، مقادیر بالای گاماگلوبولین یکباره تجویز می شود؛ اما برخی اوقات به دوز دوم دارو نیز نیاز است. در آغاز مقادیر بالای آسپیرین تا زمانی که تب وجود دارد تجویز میشود؛ بعدها میزان این دارو را کم کم کاهش میدهند. تجویز مقادیر اندک آسپیرین که اثر ضدانعقادی بر روی پلاکتها دارد ادامه می یابد تا از چسبیدن پلاکت ها به یکدیگر جلوگیری کند. هدف از این کار جلوگیری از تشکیل لخته خون(ترومبوز)در داخل آنوریسم است؛ زیرا تشکیل لخته در داخل آنوریسم می تواند باعث انفارکتوس قلبی شود که خطرناکترین عارضه بیماری کاوازاکی است.
آسپیرین در بیمارانی که ناهنجاری های عروق کرونر ندارند، برای چند هفته تجویز میشود؛ اما در کودکانی که به آنوریسم مبتلا شده اند باید برای مدت طولانی تری تجویز گردد.
پیش آگهی
جدیترین تظاهر بیماری کاوازاکی درگیری قلبی است که احتمال عارضه دار شدن آن در گذر زمان وجود دارد. صداهای غیرطبیعی قلبی، اختلالات ریتم قلبی و ناهنجاری قابل مشاهده در اکوکاردیوگرافی قلبی ممکن است وجود داشته باشد. همه لایههای مختلف قلب ممکن است تا حدودی دچار التهاب شوند. پریکاردیت (التهاب پرده های اطراف قلب)، میوکاردیت (التهاب عضله قلب)و درگیری دریچه های قلبی می تواند رخ بدهد. با این حال، تظاهر اصلی این بیماری ایجاد آنوریسم عروق کرونر است.پیش آگهی در اکثر بیماران عالی است و کودک به یک زندگی طبیعی و رشد وتکامل خوب باز میگردد .پیش آگهی بیماران مبتلا به ناهنجاری های پایدار شریانهای کرونر به میزان و شدت تنگی و انسداد که به علت تشکیل لخته های خون در داخل عروق خونی ایجاد میشود بستگی دارد.
مراقبت های لازم
توصیه میشود که بیمار برای مدت 3 تا 6 ماه از انجام واکسیناسیون خودداری کند؛ زیرا درمان با گاماگلوبولین سیستم ایمنی بدن را تحت تأثیر قرار می دهد که این اثر می تواند تا 6 ماه باقی بماند.
کودکانی که دچار درگیری قلبی نشده اند، هیچ منعی در انجام فعالیتهای ورزشی یا بدنی روزانه خود ندارند. با این حال، کودکان مبتلا به آنوریسم عروق کرونر برای شرکت در مسابقات رقابتی در سنین نوجوانی باید با یک متخصص قلب کودکان مشاوره کنند.
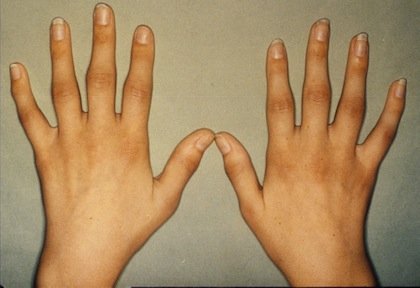
 میزان وقوع بیماری در ایالات متحده 1در 6000 تا 1در 55000 تولد زنده و در کشور فنلاند 1 در 1000تولد زنده استعلت بیماری این اختلال،ارثی بوده و علت آن ناشناخته است. دو نوع از این بیماری وجود داردکه نوع اول اتوزمال غالب است که عمدتا در بزرگ سالی بروز پیدا میکند و نوع دوم نیز اتوزومال مغلوب که در کودکی علایم آن ظاهر میشود. این بیماری در اثر جهش در جایگاه ژنی موجود در بازوی کوتاه کروموزوم 6 ( 21P6) ایجاد شده ،علیرغم نقش یک ژن منفرد در بروز بیماری، تظاهر فنوتیپیک بیماری بسیار متغیر و حتی در افراد یک خانواده میتواند متفاوت باشد .10% موارد بیماری در اثر ج هشهای خودبخود ژنی است .علایم ناشی از بیماری مراحل اولیه وجود خون در ادرار که ممکن است فقط با بررسی میکروسکوپی قابل شناسایی باشد .
میزان وقوع بیماری در ایالات متحده 1در 6000 تا 1در 55000 تولد زنده و در کشور فنلاند 1 در 1000تولد زنده استعلت بیماری این اختلال،ارثی بوده و علت آن ناشناخته است. دو نوع از این بیماری وجود داردکه نوع اول اتوزمال غالب است که عمدتا در بزرگ سالی بروز پیدا میکند و نوع دوم نیز اتوزومال مغلوب که در کودکی علایم آن ظاهر میشود. این بیماری در اثر جهش در جایگاه ژنی موجود در بازوی کوتاه کروموزوم 6 ( 21P6) ایجاد شده ،علیرغم نقش یک ژن منفرد در بروز بیماری، تظاهر فنوتیپیک بیماری بسیار متغیر و حتی در افراد یک خانواده میتواند متفاوت باشد .10% موارد بیماری در اثر ج هشهای خودبخود ژنی است .علایم ناشی از بیماری مراحل اولیه وجود خون در ادرار که ممکن است فقط با بررسی میکروسکوپی قابل شناسایی باشد .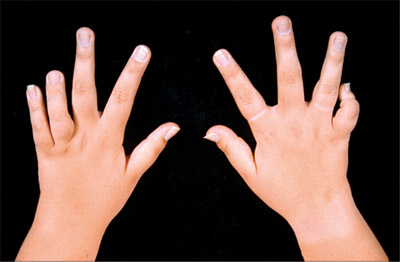
 اسامی دیگربیماری رماتیسمشیوع و همه گیری شناسی میزان شیوع این بیماری بین 03/0 تا 1/2در صد جمعیت است. زنان 3 برابر بیش از مردان مبتلا میشوند. میزان شیوع با سن افزایش می یابد و تفاوت های جنسی( در میزان شیوع) در گروه های سنی بالاتر کاهش می یابد. این بیماری در تمام مناطق جهان رخ میدهد اما به نظر میرسد شیوع بیماری در مناطق شهری بیش از روستائی است. شروع بیماری بیشتر در دهههای چهارم و پنجم زندگی است و 80% کلیه بیماران در سنین 50 -35 سالگی مبتلا به بیماری می شوند. میزان بروز آرتریت روماتوئید در زنان60- 64 ساله در مقایسه با زنان 18-29 ساله بیش از 6 برابر است. مطالعات خانوادگی دلالت بر یک استعداد ژنتیکی این بیماری میکند.علت بیماری احتمالاً به علت اختلال خود ایمنی است که طی آن دستگاه ایمنی بدن به بافت خودی حمله می کند. تورم داخل مفصل باعث تحریک انتهای اعصاب مربوط به درد می شود. آسیب به خود مفصل نیز باعث ایجاد درد می شود. وارد ساختن فشار بیش از حد به مفصل می تواند باعث افزایش تورم مفصل یا آسیب خود مفصل شده ودرد را بیشتر سازد .بسیاری از افراد به تجربه در می یابند که اگریک روز فشار بیشتری به مفاصل وارد سازند روز بعد درد بیشتری خواهند داشت. آسیب مفاصل در اثر التهاب باعث شل تر شدن رباطهامی شود. در این حالت برای آنکه استخوانها در محل خود باقی بمانند عضلات فعال تر شده و نهایتاً خستگی عضلانی بعلت فعالیت بیش از حد عضلات ایجاد میشود .علت این بیماری ناشناخته است. آرتریت مزمن در بیمارانی که دچار کمبود ایمونوگلوبولین A و گاماگلوبولین هستند رخ میدهد ،لذا ضعف سیستم دفاعی می تواند منجر به این بیماری شود. با این وجود نقص قطعی موجود نیست .فرضیه دوم این است که ممکن است ناشی از عفونت باشد گرچه هیچ گونه ارگانیسمی تا به حال شناسایی نشده است . علایم ناشی از بیماری درد، تورم ، و خشکی در مفاصل انگشتان پا ،زانو، مچ پا، آرنج ، شانه ، یا گردن . درد ممکن است به طور ناگهانی یا تدریجی آغاز شود و ممکن است(تنها در یک) یا بسیاری از مفاصل وجود داشته باشد .امکان دارد کودک بدون این که قادر به توضیح باشد، از راه رفتن امتناع کند .دمای بدن هر روز و معمولاً در عصر تا حدود 4/39درج ۀ سانتیگراد بالا میرود. تب به طور شایع با بثورات پوستی و لرز همراه است . سایر علایم عبارتند از:بی اشتهایی، کاهش وزنکم خونیتحریک پذیری، بی حالیتورم گره های لنفاویدرد و قرمزی چشمدرد قفسۀ صدری به قدری شدید است که بر قلب تأثیر می گذارد .روش تشخیص تشخیص بیماری با تکیه بر علایم بالینی و همچنین تستهای سرولوژیکی مثل الایزا و وسترن بلات و یا PCR میسر میشود .تشخیص این بیماری با مراجعه به پزشک میسر خواهد شد. البته تشخیص این بیماری راحت نیست زیرا آزمایش دقیقی جهت تشخیص این بیماری وجود ندارد، علائم این بیماری مشابه بیماریهای دیگر است و ایجاد کامل علائم، مدت زمان زیادی طول می کشد. اما جهت تشخیص مجموعه ای، از اخذ شرح حال، معاینات بالینی ،عکسبرداری و تستهای آزمایشگاهی کمک کننده می باشند.آزمایش خون، از جمله بررسی وضعی ت خود ایمنیعکس برداری از مفاصل . البته تغییرات مفصل ممکن است تا مراحل انتهایی بیماری نیز خود را در عکس مفصل نشان ندهند.درمان روان درمانی یا مشاوره برای کمک به خانواد ۀ کودک ، تا آنها بتوانند به خوبی از عهد ۀ بیماری طولانی مدت کودک برآیند. شاید مهمترین عامل دردرمان کودک حمایت عاطفی از وی باشد .جراحی برای درست کردن مفصل تغییر شکل یافته در برخی موارد مؤثر است. درمان دارویی در جهت جلوگیری از پیشرفت سریع بیماری مؤثر می باشد که شامل داروهای ضد التهاب و همچنین در برخی از بیماران داروهای سرکوب کننده سیستم ایمنی می باشد. مراقبت های لازم به هنگام حمله، کودک باید در تخت استراحت کند، مگر برای توالت ، تا زمانی که تب و سایر علایم تخفیف یابند .امکان دارد برای حفاظت و حمایت از مفصل ملتهب استفاده از آتل ضروری باشد .پس از رفع حمله ، کودک میتواند به تدریج فعالیت عادی را از سر بگیرد، البته باید در طی روز ساعت هایی را به استراحت بپردازد. کودک نباید زیاد خسته شود و هر شب باید حداقل 10-12 ساعت بخوابد .فیزیوتراپی در برخی از بیماران توصیه میشود. بعضی از حرکات را کودک خود میتواند انجام دهد ،و بعضی از آنها را والدین می توانند برای کودک انجام دهند. انجام حرکات توصیه شده مهم است زیرا کمک می کند تا درد و اثرات ناتوان کنندۀ بیماری به حداقل برسند .به دلیل تغییرات دوره ای علایم، گاهی برنامه فیزیوتراپی نیاز به بازبی نی خواهد داشت .به طور کلی از انجام ورزشهایی که بدن ک ودک در معرض ضربه قرار می گیرد باید خودداری شود، اما ک ودک باید تشویق شود تا در زمان ابتلاء به این بیماری در فعالیتهای اجتماعی شرکت کند.کودک باید یک رژیم مقوی و متعادل داشته باشد، در عین حال وی را باید تشویق به خوردن نمود.اگر تشک کودک سفت نیست ، یک تخت ۀ ضخیم چندلا زیر تشک قرار دهید .چشم ک ودک باید حداقل 2 بار در سال معاینه شود تا اگر التهابی در چشم وجود دارد زود تشخیص داده شود .بسیار مهم است که کودک به مدرسه عادی به طور روزانه برود .هر جا که لازم باشد، مدرسه باید خدمات اضافی برای برآورده کردن نیازهای کودک فراهم کند .پیش آگهی در حال حاضر قابل پیشگیری نیست . این بی ماری در حال حاضر غیر قابل درمان است . اما در عین حال، در 80%- 75% موارد ، به هنگام بلوغ یا اوایل جوانی ، بیماری به کلی فروکش کرده است .هر حمله معمولاً چند هفته به طول می انجامد و درطی دوران کودک ی، بیماری هر از چندگاهی شعله ور می شود و دوباره فروکش می کند. علایم را معمولاً می توان با درمان تحت کنترل درآورد .
اسامی دیگربیماری رماتیسمشیوع و همه گیری شناسی میزان شیوع این بیماری بین 03/0 تا 1/2در صد جمعیت است. زنان 3 برابر بیش از مردان مبتلا میشوند. میزان شیوع با سن افزایش می یابد و تفاوت های جنسی( در میزان شیوع) در گروه های سنی بالاتر کاهش می یابد. این بیماری در تمام مناطق جهان رخ میدهد اما به نظر میرسد شیوع بیماری در مناطق شهری بیش از روستائی است. شروع بیماری بیشتر در دهههای چهارم و پنجم زندگی است و 80% کلیه بیماران در سنین 50 -35 سالگی مبتلا به بیماری می شوند. میزان بروز آرتریت روماتوئید در زنان60- 64 ساله در مقایسه با زنان 18-29 ساله بیش از 6 برابر است. مطالعات خانوادگی دلالت بر یک استعداد ژنتیکی این بیماری میکند.علت بیماری احتمالاً به علت اختلال خود ایمنی است که طی آن دستگاه ایمنی بدن به بافت خودی حمله می کند. تورم داخل مفصل باعث تحریک انتهای اعصاب مربوط به درد می شود. آسیب به خود مفصل نیز باعث ایجاد درد می شود. وارد ساختن فشار بیش از حد به مفصل می تواند باعث افزایش تورم مفصل یا آسیب خود مفصل شده ودرد را بیشتر سازد .بسیاری از افراد به تجربه در می یابند که اگریک روز فشار بیشتری به مفاصل وارد سازند روز بعد درد بیشتری خواهند داشت. آسیب مفاصل در اثر التهاب باعث شل تر شدن رباطهامی شود. در این حالت برای آنکه استخوانها در محل خود باقی بمانند عضلات فعال تر شده و نهایتاً خستگی عضلانی بعلت فعالیت بیش از حد عضلات ایجاد میشود .علت این بیماری ناشناخته است. آرتریت مزمن در بیمارانی که دچار کمبود ایمونوگلوبولین A و گاماگلوبولین هستند رخ میدهد ،لذا ضعف سیستم دفاعی می تواند منجر به این بیماری شود. با این وجود نقص قطعی موجود نیست .فرضیه دوم این است که ممکن است ناشی از عفونت باشد گرچه هیچ گونه ارگانیسمی تا به حال شناسایی نشده است . علایم ناشی از بیماری درد، تورم ، و خشکی در مفاصل انگشتان پا ،زانو، مچ پا، آرنج ، شانه ، یا گردن . درد ممکن است به طور ناگهانی یا تدریجی آغاز شود و ممکن است(تنها در یک) یا بسیاری از مفاصل وجود داشته باشد .امکان دارد کودک بدون این که قادر به توضیح باشد، از راه رفتن امتناع کند .دمای بدن هر روز و معمولاً در عصر تا حدود 4/39درج ۀ سانتیگراد بالا میرود. تب به طور شایع با بثورات پوستی و لرز همراه است . سایر علایم عبارتند از:بی اشتهایی، کاهش وزنکم خونیتحریک پذیری، بی حالیتورم گره های لنفاویدرد و قرمزی چشمدرد قفسۀ صدری به قدری شدید است که بر قلب تأثیر می گذارد .روش تشخیص تشخیص بیماری با تکیه بر علایم بالینی و همچنین تستهای سرولوژیکی مثل الایزا و وسترن بلات و یا PCR میسر میشود .تشخیص این بیماری با مراجعه به پزشک میسر خواهد شد. البته تشخیص این بیماری راحت نیست زیرا آزمایش دقیقی جهت تشخیص این بیماری وجود ندارد، علائم این بیماری مشابه بیماریهای دیگر است و ایجاد کامل علائم، مدت زمان زیادی طول می کشد. اما جهت تشخیص مجموعه ای، از اخذ شرح حال، معاینات بالینی ،عکسبرداری و تستهای آزمایشگاهی کمک کننده می باشند.آزمایش خون، از جمله بررسی وضعی ت خود ایمنیعکس برداری از مفاصل . البته تغییرات مفصل ممکن است تا مراحل انتهایی بیماری نیز خود را در عکس مفصل نشان ندهند.درمان روان درمانی یا مشاوره برای کمک به خانواد ۀ کودک ، تا آنها بتوانند به خوبی از عهد ۀ بیماری طولانی مدت کودک برآیند. شاید مهمترین عامل دردرمان کودک حمایت عاطفی از وی باشد .جراحی برای درست کردن مفصل تغییر شکل یافته در برخی موارد مؤثر است. درمان دارویی در جهت جلوگیری از پیشرفت سریع بیماری مؤثر می باشد که شامل داروهای ضد التهاب و همچنین در برخی از بیماران داروهای سرکوب کننده سیستم ایمنی می باشد. مراقبت های لازم به هنگام حمله، کودک باید در تخت استراحت کند، مگر برای توالت ، تا زمانی که تب و سایر علایم تخفیف یابند .امکان دارد برای حفاظت و حمایت از مفصل ملتهب استفاده از آتل ضروری باشد .پس از رفع حمله ، کودک میتواند به تدریج فعالیت عادی را از سر بگیرد، البته باید در طی روز ساعت هایی را به استراحت بپردازد. کودک نباید زیاد خسته شود و هر شب باید حداقل 10-12 ساعت بخوابد .فیزیوتراپی در برخی از بیماران توصیه میشود. بعضی از حرکات را کودک خود میتواند انجام دهد ،و بعضی از آنها را والدین می توانند برای کودک انجام دهند. انجام حرکات توصیه شده مهم است زیرا کمک می کند تا درد و اثرات ناتوان کنندۀ بیماری به حداقل برسند .به دلیل تغییرات دوره ای علایم، گاهی برنامه فیزیوتراپی نیاز به بازبی نی خواهد داشت .به طور کلی از انجام ورزشهایی که بدن ک ودک در معرض ضربه قرار می گیرد باید خودداری شود، اما ک ودک باید تشویق شود تا در زمان ابتلاء به این بیماری در فعالیتهای اجتماعی شرکت کند.کودک باید یک رژیم مقوی و متعادل داشته باشد، در عین حال وی را باید تشویق به خوردن نمود.اگر تشک کودک سفت نیست ، یک تخت ۀ ضخیم چندلا زیر تشک قرار دهید .چشم ک ودک باید حداقل 2 بار در سال معاینه شود تا اگر التهابی در چشم وجود دارد زود تشخیص داده شود .بسیار مهم است که کودک به مدرسه عادی به طور روزانه برود .هر جا که لازم باشد، مدرسه باید خدمات اضافی برای برآورده کردن نیازهای کودک فراهم کند .پیش آگهی در حال حاضر قابل پیشگیری نیست . این بی ماری در حال حاضر غیر قابل درمان است . اما در عین حال، در 80%- 75% موارد ، به هنگام بلوغ یا اوایل جوانی ، بیماری به کلی فروکش کرده است .هر حمله معمولاً چند هفته به طول می انجامد و درطی دوران کودک ی، بیماری هر از چندگاهی شعله ور می شود و دوباره فروکش می کند. علایم را معمولاً می توان با درمان تحت کنترل درآورد . 
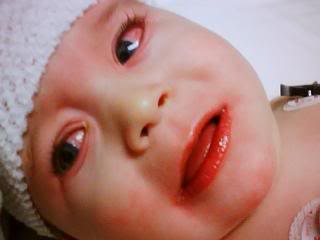
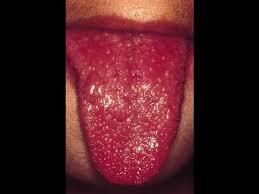
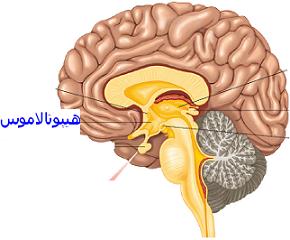
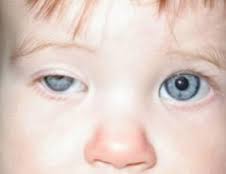
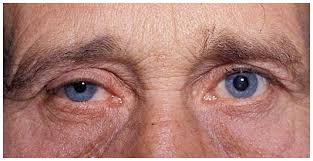 سندرم هورنر ،اختلالی است که به ندرت جمعیت عمومی با آن مواجه میشوند و شاید در هر شرایط سنی رخ دهد، شیوع بیماری در هر دو جنس یکسان است. در جمعیت مورد مطالعه شیوع این بیماری 421 در 100000 نفر در افراد زیر 19 سال و 1 در 6250نفر در حاملین این بیماری تخمین زده شده است.علت بیماری سندرم هورنر یک اختلال عصبی که حاصل قطع شدن عصب فیبر که از قسمت مغز تا صورت و چشمها ادامه دارد می باشد ،شرایط متفاوتی منجر به ایجاد این وضعیت می شوند و علل آن شامل: نامشخص، ضربه، تومور مغزی ،میگرن، دردهای خوشه ای سر، پرکاری تیروئید در سطع وسیع، سرطان ریه.علایم ناشی از بیماری علائم بیماری از سندرم هورنر به صورت خاص است یعنی یک مجموعه سه تائی از علائمی که یک طرف از صورت را تحت تأثیر قرار می دهد سه علائم مذبور عبارتند از: کم شدن عرق، افتادگی یک پلک، تنگی حدقه چشم. ارزیابی بالینی از افراد مبتلا برای شناسائی وضع موجود مناسب هستند. آزمایشات نورولوژی و شرح حال شاید در تأیید تشخیص این بیماری کمک کنند. سی تی اسِکن قفسه سینه، سی تی اسِکن مغز، رگ نگاری مغزی، نمونه برداری مایع نخاعی شاید برای بررسی سندرم هورنر انجام گیرد.روش تشخیص تشخیص به صورت بالینی بوده و از روی علائم و نشانه ها قابل تشخیص است. ممکن است استفاده از تصویر برداری و یا نوار عصبی و عضلانی برای افزایش اطلاعات و تایید نهایی استفاده شود.درمان نوع اولیه که علت مشخص ندارد تا به امروز درمان نداشته اما در انواع ثانویه و ثالثیه درمان در شرایط اکتسابی به سوی ریشه کنی علت به وجود آورنده بیماری سوق داده میشود . مراقبت های لازمحمایت روحی و روانی بیمار لازم می باشد. در خصوص هریک از مشکلات به وجود آمده نیز درمان مربوطه صورت میگیرد. از آنجایکه درمان قطعی بیماری تا به حال میسر نمی باشد لذا استفاد از تجهیزات کمی در جهت افزایش سطح زندگی بیمار مهم است.پیش آگهیپیش آگهی این بیماری به علت زمینه ای سندروم بستگی دارد. در صورت مادرزادی بودن امکان درمان قطعی وجود ندارد ولی اگر بصورت زمینه ای باشد معمولا با رفع علل به وجود آورنده علایم بیماری نیز تقلیل می یابد.
سندرم هورنر ،اختلالی است که به ندرت جمعیت عمومی با آن مواجه میشوند و شاید در هر شرایط سنی رخ دهد، شیوع بیماری در هر دو جنس یکسان است. در جمعیت مورد مطالعه شیوع این بیماری 421 در 100000 نفر در افراد زیر 19 سال و 1 در 6250نفر در حاملین این بیماری تخمین زده شده است.علت بیماری سندرم هورنر یک اختلال عصبی که حاصل قطع شدن عصب فیبر که از قسمت مغز تا صورت و چشمها ادامه دارد می باشد ،شرایط متفاوتی منجر به ایجاد این وضعیت می شوند و علل آن شامل: نامشخص، ضربه، تومور مغزی ،میگرن، دردهای خوشه ای سر، پرکاری تیروئید در سطع وسیع، سرطان ریه.علایم ناشی از بیماری علائم بیماری از سندرم هورنر به صورت خاص است یعنی یک مجموعه سه تائی از علائمی که یک طرف از صورت را تحت تأثیر قرار می دهد سه علائم مذبور عبارتند از: کم شدن عرق، افتادگی یک پلک، تنگی حدقه چشم. ارزیابی بالینی از افراد مبتلا برای شناسائی وضع موجود مناسب هستند. آزمایشات نورولوژی و شرح حال شاید در تأیید تشخیص این بیماری کمک کنند. سی تی اسِکن قفسه سینه، سی تی اسِکن مغز، رگ نگاری مغزی، نمونه برداری مایع نخاعی شاید برای بررسی سندرم هورنر انجام گیرد.روش تشخیص تشخیص به صورت بالینی بوده و از روی علائم و نشانه ها قابل تشخیص است. ممکن است استفاده از تصویر برداری و یا نوار عصبی و عضلانی برای افزایش اطلاعات و تایید نهایی استفاده شود.درمان نوع اولیه که علت مشخص ندارد تا به امروز درمان نداشته اما در انواع ثانویه و ثالثیه درمان در شرایط اکتسابی به سوی ریشه کنی علت به وجود آورنده بیماری سوق داده میشود . مراقبت های لازمحمایت روحی و روانی بیمار لازم می باشد. در خصوص هریک از مشکلات به وجود آمده نیز درمان مربوطه صورت میگیرد. از آنجایکه درمان قطعی بیماری تا به حال میسر نمی باشد لذا استفاد از تجهیزات کمی در جهت افزایش سطح زندگی بیمار مهم است.پیش آگهیپیش آگهی این بیماری به علت زمینه ای سندروم بستگی دارد. در صورت مادرزادی بودن امکان درمان قطعی وجود ندارد ولی اگر بصورت زمینه ای باشد معمولا با رفع علل به وجود آورنده علایم بیماری نیز تقلیل می یابد. 


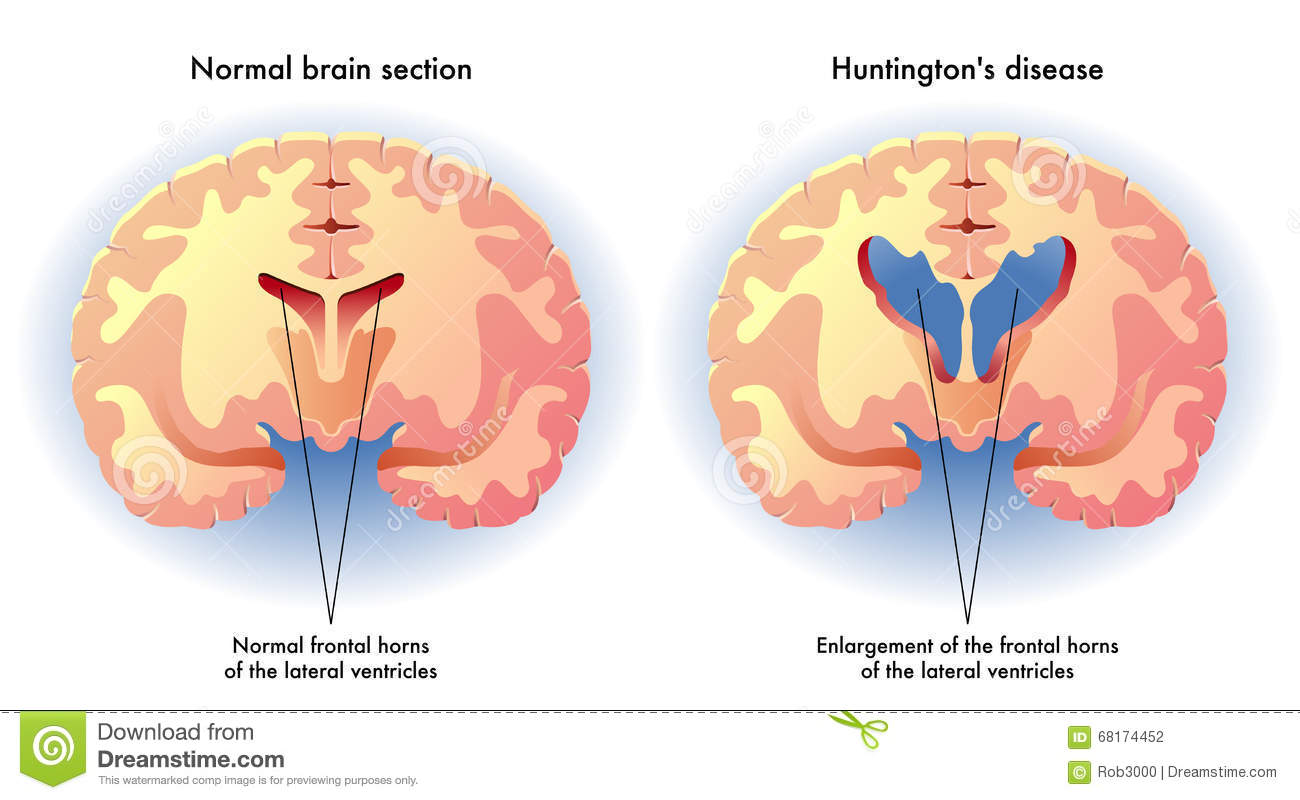 بروز این بیماری 2 تا 6 مورد از هر 000،100 نفر در سال میباشد میزان بروز در مردان و زنان برابر است. تقریبا 80 درصد بیماران نوجوان، ژن HD جهش یافته را از پدر به ارث می برند. تقریبا 3/1بیماران با اختلالات روانی تظاهر میکنند و دو سوم بیماران با ترکیبی از اختلالات شناختی و حرکتی مراجعه می کنند. علت بیماری هانتینگتون، نوعی اختلال تحلیل برنده عصبی اتوزومی غالب است که در تمام نژادها به چشم می خورد و به علت جهشهایی در ژن HD ایجاد میشود. این ژن حاوی یک توالی تکرارشونده سه نوکلئوتیدی(CAG)است که برای تشخیص بیماری هانتینگتون به کار میرود. در این بیماری هسته دم دار تالاموس، ساقه مغز و قسمت های دیگری از مغز آتروفی میگردد .علایم ناشی از بیماری هانتینگتون با اختلالات پیشرونده حرکتی، شناختی و روانی مشخص می شود. اختلالات حرکتی شامل حرکات ارادی و نیز غیرارادی می شوند. در ابتدا این حرکات به میزان ناچیزی در فعالیتهای روزانه مداخله می کنند، اما با پیشرفت بیماری، عموما ناتوان کننده می شوند. علایم مشخصه این بیماری ،تکان ها و حرکات غیرعادی، اختلال در تکلم، اختلال در تعقل و حالات عاطفی است. علایم بیماری عبارتند از:تحلیل رفتن جسمی و روانیبی حالی و بیتفاوتی(آپاتی)افسردگی یا هیجان پذیریاختلال حسیپیچ و تاب بی اراده دست و پاعدم تعادلحرکات ناموزون صورتچرخاندن زیاد لب به دور دهانصدا درآوردن با لب هاصحبت کردن غیرعادی همراه با فشار و صداهای انفجار آمیز.روش تشخیص یافته های حاصل اسکن مغز نشان دهنده تحلیل بافتی در مغز بوده و تصویربرداری با ام. ار.ای نیز کمک کننده است. آزمایشات ژنتیکی تایید کننده نهایی بیماری می باشد .درمان در حال حاضر ،هیچ درمان علاج دهنده ای برای بیماری هانتینگتون وجود ندارد. درمان بر مراقبت حمایتی و نیز اداره دارویی مشکلات رفتاری و عصبی متمرکز است. با پیشرفت کره بستری کردن در آسایشگاهها لازم میشود. علایم بیخوابی، اضطراب، و افسردگی را میتوان با مصرف بنزودیازپینها و ضدافسردگیها رفع کرد. علایم عصبی را میتوان با داروهای آنتی سایکوتیک پرقدرت یا آنتاگونیستهای سروتونین، دوپامین درمان کرد. مشاوره ژنتیک مهمترین مداخله میباشد .مراقبت های لازم افسردگی شدید یکی از مشکلات عمده در بیماران می باشد که نیازمند توجه بیشتر به بیمار است. بیمار در معرض ناتوانی جسمی برای انجام کارهای روزمره، احتمال آسیب به خود یا دیگران، افزایش خطر عفونت می باشد . پیش آگهی سیر این بیماری پیشرونده بوده و معمولاً 15 تا 20 سال بعد از تشخیص منجر به مرگ بیمار می شود. خودکشی در این بیماری شایع است.
بروز این بیماری 2 تا 6 مورد از هر 000،100 نفر در سال میباشد میزان بروز در مردان و زنان برابر است. تقریبا 80 درصد بیماران نوجوان، ژن HD جهش یافته را از پدر به ارث می برند. تقریبا 3/1بیماران با اختلالات روانی تظاهر میکنند و دو سوم بیماران با ترکیبی از اختلالات شناختی و حرکتی مراجعه می کنند. علت بیماری هانتینگتون، نوعی اختلال تحلیل برنده عصبی اتوزومی غالب است که در تمام نژادها به چشم می خورد و به علت جهشهایی در ژن HD ایجاد میشود. این ژن حاوی یک توالی تکرارشونده سه نوکلئوتیدی(CAG)است که برای تشخیص بیماری هانتینگتون به کار میرود. در این بیماری هسته دم دار تالاموس، ساقه مغز و قسمت های دیگری از مغز آتروفی میگردد .علایم ناشی از بیماری هانتینگتون با اختلالات پیشرونده حرکتی، شناختی و روانی مشخص می شود. اختلالات حرکتی شامل حرکات ارادی و نیز غیرارادی می شوند. در ابتدا این حرکات به میزان ناچیزی در فعالیتهای روزانه مداخله می کنند، اما با پیشرفت بیماری، عموما ناتوان کننده می شوند. علایم مشخصه این بیماری ،تکان ها و حرکات غیرعادی، اختلال در تکلم، اختلال در تعقل و حالات عاطفی است. علایم بیماری عبارتند از:تحلیل رفتن جسمی و روانیبی حالی و بیتفاوتی(آپاتی)افسردگی یا هیجان پذیریاختلال حسیپیچ و تاب بی اراده دست و پاعدم تعادلحرکات ناموزون صورتچرخاندن زیاد لب به دور دهانصدا درآوردن با لب هاصحبت کردن غیرعادی همراه با فشار و صداهای انفجار آمیز.روش تشخیص یافته های حاصل اسکن مغز نشان دهنده تحلیل بافتی در مغز بوده و تصویربرداری با ام. ار.ای نیز کمک کننده است. آزمایشات ژنتیکی تایید کننده نهایی بیماری می باشد .درمان در حال حاضر ،هیچ درمان علاج دهنده ای برای بیماری هانتینگتون وجود ندارد. درمان بر مراقبت حمایتی و نیز اداره دارویی مشکلات رفتاری و عصبی متمرکز است. با پیشرفت کره بستری کردن در آسایشگاهها لازم میشود. علایم بیخوابی، اضطراب، و افسردگی را میتوان با مصرف بنزودیازپینها و ضدافسردگیها رفع کرد. علایم عصبی را میتوان با داروهای آنتی سایکوتیک پرقدرت یا آنتاگونیستهای سروتونین، دوپامین درمان کرد. مشاوره ژنتیک مهمترین مداخله میباشد .مراقبت های لازم افسردگی شدید یکی از مشکلات عمده در بیماران می باشد که نیازمند توجه بیشتر به بیمار است. بیمار در معرض ناتوانی جسمی برای انجام کارهای روزمره، احتمال آسیب به خود یا دیگران، افزایش خطر عفونت می باشد . پیش آگهی سیر این بیماری پیشرونده بوده و معمولاً 15 تا 20 سال بعد از تشخیص منجر به مرگ بیمار می شود. خودکشی در این بیماری شایع است.
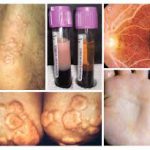 شیوع و همه گیر شناسی نزدیک به 5% از کل جمعیت از لحاظ 2E2/apoE هوموزیگوت هستند اما فقط بخش کوچکی از این افراد دچار این بیماری میشوند .علت بیماری نقص ژنتیکی باعث این بیماری است. این نقص از تجمع ذرات بزرگ لیپوپروتئینی که حاوی کلسترول و تری گلیسرید که خود نوعی چربی است به وجود می آید. این بیماری با نقص در ژن آپولیپو پروتئین E در ارتباط است. در بیشتر موارد، یک عامل اضافی شناسایی دیگر در بروز هیپرلیپوپروتئینمی دخیل است. شایع ترین این عوامل رژیم های پرکالری – پر چربی، دیابت، چاقی ،هیپرلیپیدمی می باشند که در بیشتر موارد هیپرلیپیدمی فامیلی و مرکب(FCHL)یا هیپرکلسترولمی فامیلی(FH)است. جهش های نادری در apoE باعث شک لگیری اشکال غالب FDBL می شوند؛ در این موارد هیپرلیپیدمی به طور کامل، در شرایط هتروزیگوت، بروز می نماید.علایم ناشی از بیماری بیماران دچار FDBL معمولاً در جوانی دچار گزانتوم می شوند و بیماری های عروق کرونری و محیطی زودرس پیدا می کنند بیماری به ندرت در زنان ،پیش از یائسگی بروز می کند. دو نوع ششخص از گزانتوم ها در بیماران دچار FDBL دیده میشوند: –Tubero eruptive و گزانتوم های کف دست. گزانتوم هایی که به صورت خوشه هایی از ضایعات پوستی برجسته روی آرنجها زانوها یا باسن ها شروع شده و میتوانند به اندازۀ دانههای کوچک انگور برسند. گزانتوم کف دست(که به آن استریاتاپالماریس هم گفته می شود)تغییر رنگهای نارنجی مایل به زرد کف دست هستند.روش تشخیصرویکرد کلاسیک، تشخیص این اختلال استفاده از الکتروفورز لیپوپروتئین ها است؛ در FDBL، لیپوپروتئی نهای باقی مانده به صورت یک باند β پهن تجمع می کنند، روش ارجح برای تأیید تشخیص FDBL اندازهگیری VLDL- 3با استفاده از اولتراسانتریفوژ و مشخص کردن نسبت VLDL-C به کل تری گلیسرید پلاسما است؛ نسبت 30%> نشاندهندۀ FDBL است. آزمایشاتی که ممکن است برای تشخیص این بیماری انجام شود عبارتند از:تست ژنتیکی برای آپولیپو پروتئیناسترس تست قلبکلسترول تامتری گلیسیریدتست( VLDL)لیپوپروتئینی با حجم بسیار کمدرمان افراد دچار FCHL را باید به صورت تهاجمی درمان کرد چرا که خطر CHD زودرس در آنها به طور مشخص بالا است .کاهش مصرف خوراکی چربی اشباع و کربوهیدرات ساده، ورزش هوازی و کاهش وزن اثرات مفیدی روی پروفایل لیپید دارد. بیماران دیابتی را باید به صورت تهاجمی درمان کرد تا سطح گلوکز خون در حد مطلوب بماند. بیشتر بیماران FCHL نیاز به داروهای ضد چربی دارند تا لیپوپروتئین هایشان به حد مطلوب برسد. در زنان یائسه، دیس لیپیدمی به جایگزینی هورمونی استروژن پاسخ میدهد و درمان دارویی ترکیبی گاهی اوقات مورد نیاز خواهد بود.مراقبت های لازم درمان زود هنگام و اجتناب از سایر عوامل خطرساز برای بیماری های عروقی )مانند سیگار کشیدن( برای جلوگیری از حملات قلبی زودرس، سکته مغزی و مسدود شدن رگهای خونی بسیار مهم و ضروری هستند.پیش آگهی سایر شرایط متابولیک که می توانند هیپرلیپیدمی را بدتر کنند باید فعالانه درمان شوند. بیماران FDBL به طور تیپیک خیلی خوب به رژیم غذایی پاسخ میدهند کاهش وزن و رژیم های کم کلسترول کم چربی میتواند پاسخ خوب در این بیماران ایجاد نمایند .مصرف الکل باید قطع شود .پیش آگهی به پایبندی و پاسخ فردی به برنامۀ درمانی بستگی دارد. هدف درمان پیشگیری از عوارض ثانویه آن از جمله بی ماریهای قلبی و عروقی است.
شیوع و همه گیر شناسی نزدیک به 5% از کل جمعیت از لحاظ 2E2/apoE هوموزیگوت هستند اما فقط بخش کوچکی از این افراد دچار این بیماری میشوند .علت بیماری نقص ژنتیکی باعث این بیماری است. این نقص از تجمع ذرات بزرگ لیپوپروتئینی که حاوی کلسترول و تری گلیسرید که خود نوعی چربی است به وجود می آید. این بیماری با نقص در ژن آپولیپو پروتئین E در ارتباط است. در بیشتر موارد، یک عامل اضافی شناسایی دیگر در بروز هیپرلیپوپروتئینمی دخیل است. شایع ترین این عوامل رژیم های پرکالری – پر چربی، دیابت، چاقی ،هیپرلیپیدمی می باشند که در بیشتر موارد هیپرلیپیدمی فامیلی و مرکب(FCHL)یا هیپرکلسترولمی فامیلی(FH)است. جهش های نادری در apoE باعث شک لگیری اشکال غالب FDBL می شوند؛ در این موارد هیپرلیپیدمی به طور کامل، در شرایط هتروزیگوت، بروز می نماید.علایم ناشی از بیماری بیماران دچار FDBL معمولاً در جوانی دچار گزانتوم می شوند و بیماری های عروق کرونری و محیطی زودرس پیدا می کنند بیماری به ندرت در زنان ،پیش از یائسگی بروز می کند. دو نوع ششخص از گزانتوم ها در بیماران دچار FDBL دیده میشوند: –Tubero eruptive و گزانتوم های کف دست. گزانتوم هایی که به صورت خوشه هایی از ضایعات پوستی برجسته روی آرنجها زانوها یا باسن ها شروع شده و میتوانند به اندازۀ دانههای کوچک انگور برسند. گزانتوم کف دست(که به آن استریاتاپالماریس هم گفته می شود)تغییر رنگهای نارنجی مایل به زرد کف دست هستند.روش تشخیصرویکرد کلاسیک، تشخیص این اختلال استفاده از الکتروفورز لیپوپروتئین ها است؛ در FDBL، لیپوپروتئی نهای باقی مانده به صورت یک باند β پهن تجمع می کنند، روش ارجح برای تأیید تشخیص FDBL اندازهگیری VLDL- 3با استفاده از اولتراسانتریفوژ و مشخص کردن نسبت VLDL-C به کل تری گلیسرید پلاسما است؛ نسبت 30%> نشاندهندۀ FDBL است. آزمایشاتی که ممکن است برای تشخیص این بیماری انجام شود عبارتند از:تست ژنتیکی برای آپولیپو پروتئیناسترس تست قلبکلسترول تامتری گلیسیریدتست( VLDL)لیپوپروتئینی با حجم بسیار کمدرمان افراد دچار FCHL را باید به صورت تهاجمی درمان کرد چرا که خطر CHD زودرس در آنها به طور مشخص بالا است .کاهش مصرف خوراکی چربی اشباع و کربوهیدرات ساده، ورزش هوازی و کاهش وزن اثرات مفیدی روی پروفایل لیپید دارد. بیماران دیابتی را باید به صورت تهاجمی درمان کرد تا سطح گلوکز خون در حد مطلوب بماند. بیشتر بیماران FCHL نیاز به داروهای ضد چربی دارند تا لیپوپروتئین هایشان به حد مطلوب برسد. در زنان یائسه، دیس لیپیدمی به جایگزینی هورمونی استروژن پاسخ میدهد و درمان دارویی ترکیبی گاهی اوقات مورد نیاز خواهد بود.مراقبت های لازم درمان زود هنگام و اجتناب از سایر عوامل خطرساز برای بیماری های عروقی )مانند سیگار کشیدن( برای جلوگیری از حملات قلبی زودرس، سکته مغزی و مسدود شدن رگهای خونی بسیار مهم و ضروری هستند.پیش آگهی سایر شرایط متابولیک که می توانند هیپرلیپیدمی را بدتر کنند باید فعالانه درمان شوند. بیماران FDBL به طور تیپیک خیلی خوب به رژیم غذایی پاسخ میدهند کاهش وزن و رژیم های کم کلسترول کم چربی میتواند پاسخ خوب در این بیماران ایجاد نمایند .مصرف الکل باید قطع شود .پیش آگهی به پایبندی و پاسخ فردی به برنامۀ درمانی بستگی دارد. هدف درمان پیشگیری از عوارض ثانویه آن از جمله بی ماریهای قلبی و عروقی است.