
در ناامیدی بسی امیداست پایان شب سیه سفیداست
[می خواهم ثابت کنم فرزندانم می توانند آینده خوبی داشته باشند
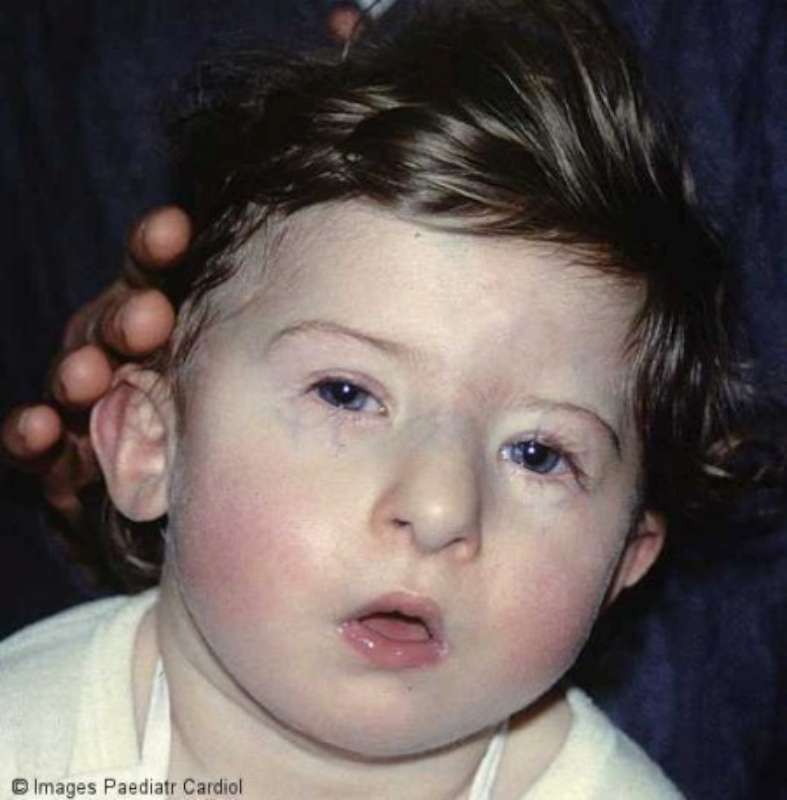
آقای ذاکری و همسرش بعداز اینکه از پزشکان ایرانی ناامید شدند دنبال راه چاره گشتند. وی در این خصوص گفت : با همسرم تصمیم گرفتیم به دنبال راه چاره بگردیم. ما باید دارویی پیدا میکردیم که به فرزندانمان کمک می کرد. با دکتر زمان در این خصوص مشورت کردیم و به او گفتیم به دنبال پیوند استخوان هستیم . ایشان گفتند بیماری فرزندان ما به گونه ای است که نمی توان به آنها آنزیم تزریق کرد. پسرانم مشکلشان جدی شده بود و به شدت عصبی می شدند. بیماری داشت شدت پیدا می کرد و باید زودتر راه چاره ای پیدا می کردیم. وقتی دکتر زمان از ایران رفت از طریق ایمیل با او در ارتباط بودیم. به او پیشنهاد دادیم که پیوند مغز و استخوان انجام دهیم اما او گفت : این روش فایده ای ندارد اما دارویی به اسم “جینستین” هست که می تواند به درمان یا ماوقف سازی این بیماری کمک کند. از اینجا بود که کار دشوار آقای ذاکری و همسرش شروع شد. آنها باید به دنبال راه حلی برای پیداکردن این دارو می گشتند تا بتوانند به فرزندانشان کمک کنند. ذاکری درباره ی پیدا کردن این دارو می گوید : ما اولین خانواده در ایران هستیم که توانستیم این دارو را پیدا کنیم. توانستیم با یک پروفسور آلمانی آشنا شویم واو به ما کمک زیادی کرد. این پروفسور گفت که توانسته پودری را از مشقات سویا تهیه کند که برای درمان این بیماری مهم است. او گفت آزمایش هایی رو روی موش آزمایشگاهیی انجام داده و نتیجه مثبت بوده است. این پودر اثرات بازدارنده مثبتی داشته است و می تواند عوارض این بیماری را متوقف کند اما مشکل ما این بود که این دارو را باید از کجا تهیه می کردیم ؟ بعد از جست وجو و تلاش زیاد فهمیدیم که این دارو را می توانیم از یک داروخونه در نیویورک تهیه کنیم و تنها در آنجا این پودر موجود است. الان هم نزدیک به چند ماه است که با تمام سختی ها و مشکلات این پودر را تهیه می کنیم.وی و همسرش مرارت های زیادی را برای پیدا کردن راه معالجه پسرانشان تحمل کرده اند و در این راه بار ها ناامید شده اند اما هر بار با عزم و اراده ای راسخ تر به دنبال راه چاره ای برای بهبودی و یا حداقل متوقف سازی بیماری فرزندانشان گشته اند …از این پدر فداکار میپرسیم از کی متوجه شده اند که فرزندانشان با چنین بیماری نادری دست وپنجه گرم می کنند.؟ وی پاسخ می دهد :از دوران کودکی پسرم شب ها نمی خوابید. گریه های شدیدی می کرد و به هر دکتری هم مراجعه می کردیم او نمی توانست راه چاره ای به ما نشان دهد تا اینکه همانطور که توضیح دادم فهمیدیم نا این بیماری MTS است. پایه و اساس این بیماری استرس و بی قراری است که به مرور زمان پیشرفت می کند. این بیماری روی استخوان، مغز و کبد تاثیر می گذارد.ما حتی فرزندانمان را پیش روانپزشک کودک هم بردیم که آنها فقط داروی اعصاب تجویز کردند. الان دیگر پسرانم آن دارو ها را مصرف نمی کنند و فقط همان پودر را می خورند. خوشبختانه حالشان بهتر شده و درد استخوانشان هم کمتر شده است. ذاکری درباره اینکه از مسئولان چه انتظاراتی درباره چنین بیماری های نادری دارندد، گفت : وقتی پودر “جینسیتین ” را پیدا کردیم به بنیاد بیماری های خاص رفتیم و گفتیم چنین پودری هست که می تواند به متوقف سازی این بیماری کمک کند اما آنجا کسی حرف ما را جدی نگرفت و گفتند خوتان را اذیت نکنید. آنها حتی حاضر نشدند در اینترنت راجع به این دارو جست وجو کنند. ما حتی خواستیم شماره تلفن خانواده هایی که با این بیماری روبرو هستند را به ما بدهند تا به آنها هم کمک کنیم، اما این کار را هم نکردند. در حال حاضر هم تهیه این دارو برای ما خیلی دشوار است. علاوه بر تامین هزینه آن که مبلغی در حدود 800هزار تومان در هر ماه میشود باید یکی راهم پیدا کنیم که در رفت وآمد از ایران به آمریکا باشد و بتوانند دارو را برای ما بیاورد. وی اضافه کرد : در کنار استفاده از این دارو فرزندانم را به انرژی درمانی می بریم و در آنجا فردی هست که بدون دریافت هزینه به ما کمک میکند تا این ثمرات زندگیمان را دوباره صحیح و سالم کنار خود ببینیم. آقای ذاکری در ادامه صحبت هایش می گوید : از وقتی با بنیاد بیماری های نادر آشنا شده احساس می کند ،پشتش به جایی گرم است؛ وی در این خصوص گفت : وقتی به بنیاد بیماری های نادر مراجعه کردم تفکرم نسبت به آنها متفتوت بود و ذهنیت خوبی نداشتم اما حالا احساس می کنم پشتم به جایی گرم است. این بنیاد کارتی برای ما صادر کرد و اواخر اسفندماه هم تماس گرفتند و مبلغی را به حسابمان واریز کردند که پول یکماه پودر بچه هایم بود. اگرچه بعد مالی هم مهم است اما با عضویت در این بنیاد دیگر احساس تنهایی و ناامیدی نمی کنیم. فردی که این دارو را برای ما می آورد دکتر حسین غفوریان است که در آمریکا چشم پزشکی می خواند اما حالا به ایران برگشته وما به دنبال راه دیگری برای تهیه دارو هستیم و از بنیاد می خواهم در این خصوص کمکمان کند. بااین حال فکر می کنم با پیدا کردن این دارو معجزه شد وخدا کمکمان کرد. وی در خاتمه گفت : از مسئولین بنیادبیماری های نادر تشکر میکنم که مارا در سخترین لحظات تنها نمیگذارند و در نهایت می گویم که “میخواهم به خیلی ها ثابت کنم پسرانم می توانند آینده خوبی داشته باشند”.

 بدون هیچ وسیله نقلیه ای برای حمل و نقل فرزندانشان مدت 26 سال است که زندگی استیجاری خود را در کنار این همه در دو غم جانکاه و مشکلات عدیده دیگر که همه را تحت الشعاع قرار داده سپری می کنند چرا که ایمان مطلق به خدا و اعتقادات قبلی و امید به زندگی و آینده و این که قضاء و قدر برای عده ای از بندگان بصورت ویژه قرار گرفته و دسترنج آن را خواهند دید، صبر ایوب در پیش گرفته و راضی به رضای خداوند منان هستند. بقول معروف کجا خوش است آنجا که دل خوش است و حتی خداوند به خانواده آقا سید مهدی می گوید: ما نباید درهایی را که خدا بسوی ما می گشاید ،ندانسته ببندیم و درهایی را که برویمان می بندد به اصرار بگشاییم چون حکمت و مصلحت بندگان را فقط خالق دانا می داند و بس.
مبدا همه نیاز ها نعمت سلامتی و عافیت و مقصد همه این نیازها عاقبت به چیزی است مابین این مبدا ال آن مقصد والاترین و بهترین نیاز ها عاقبت به چیزی است ما بین این مبدا تا آن مقصد والاترین و بهترین نیازها دلخوشی است. به حق عظمت خداوندی آنرا برای همه شما عزیزان و خانواده هایتان و همه خوانندگان و دوستاران بشر آرزو مندیم.
بدون هیچ وسیله نقلیه ای برای حمل و نقل فرزندانشان مدت 26 سال است که زندگی استیجاری خود را در کنار این همه در دو غم جانکاه و مشکلات عدیده دیگر که همه را تحت الشعاع قرار داده سپری می کنند چرا که ایمان مطلق به خدا و اعتقادات قبلی و امید به زندگی و آینده و این که قضاء و قدر برای عده ای از بندگان بصورت ویژه قرار گرفته و دسترنج آن را خواهند دید، صبر ایوب در پیش گرفته و راضی به رضای خداوند منان هستند. بقول معروف کجا خوش است آنجا که دل خوش است و حتی خداوند به خانواده آقا سید مهدی می گوید: ما نباید درهایی را که خدا بسوی ما می گشاید ،ندانسته ببندیم و درهایی را که برویمان می بندد به اصرار بگشاییم چون حکمت و مصلحت بندگان را فقط خالق دانا می داند و بس.
مبدا همه نیاز ها نعمت سلامتی و عافیت و مقصد همه این نیازها عاقبت به چیزی است مابین این مبدا ال آن مقصد والاترین و بهترین نیاز ها عاقبت به چیزی است ما بین این مبدا تا آن مقصد والاترین و بهترین نیازها دلخوشی است. به حق عظمت خداوندی آنرا برای همه شما عزیزان و خانواده هایتان و همه خوانندگان و دوستاران بشر آرزو مندیم.



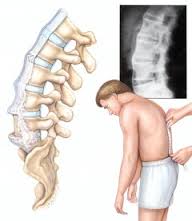
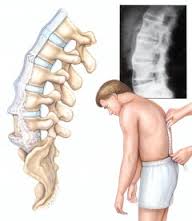 مشخص ترین علامت این بیماری،درد ستون فقرات ولگن است و از آنجا که درد کمری، یکی از شایع ترین شکایات افراد هر جامعه است، تعداد زیادی از کسانی که مبتلا به این بیماری می شوند، ممکن است با تشخیص های دیگری درمان شوند.بسیاری از بیماران هستند که ماه ها یا سال ها قبل از اینکه به طور قطعی، تشخیص بیماری یشان داده شود، در بخش های مختلف داخل کشور، سرگردان بوده وبرای تعداد دیگری از بیماران نیز بررسی ها،تشخیص ودرمان اشتباه صورت میگیرد که این، خود عاملی است برای اضافه شدن اختلالات دیگری در ستون فقرات که درنهایت، درد ناشی از اقدامات صورت گرفته، به درد کمری و لگن اضافه می شودومشکل بیماری را برحسب وسعت اقداماتی که انجام گرفته، بیشتر می کند.درد ناشی از AS اکثرابه آهستگی و خود به خود شروع می شود و ماه ها به طور ثابت یا پیش رونده باقی می ماند.این درد با استراحت، بیشتر می شود وبا فعالیت کاهش می یابد.درد لگن وستون فقرات پشتی-کمری،اکثرا بیمار رادر نیمه دوم شب از خواب بیدار شده وبرحسب شدت درد ، مدتی راه رفته و دوباره به رختخواب برگردد.وقتی صبحگاهان فرا می رسد وبیمار،از خواب بلند می شود،درد بیشتر است وخشکی فقرات کمری ولگن وجود دارد.هرچه بیماری شدیدتر باشد، خشکی صبحگاهی بیشتر طول میکشد وگاهی در بعضی از بیماران تا نیمه روز، خشکی باقی می ماند.وقتی بیمار از رختخواب بیرون می آید،خشکی صبحگاهی و دردشدید وجود دارد ولی با شروع فعالیت، درد وخشکی، به تدریج از بین رفته واکثرا،بیمار، آن روزها احساس راحتی می کند؛در عده کمی از مبتلایان به طور کلی درد وجودندارد.یکی از خصوصیات این بیماری،التهاب محل چسبیدن رباط ها به استخوان است، لذا هر جایی که رباط ها به استخوان می چسبند،بیماران، احساس درد می کنند، مثلا درد در پاشنه ها ودرد قفسه صدری وجود داردویا حتی ممکن است در بعضی ها، شروع بیماری با درد پاشنه ها وقفسه صدری باشد.علاوه برگرفتاری مفاصل لگن،ستون فقرات،پاشنه هاوقفسه صدری ومحل چسبیدن رباط ها به استخوان ها،در این بیماری،مفاصل بزرگ اندام ها نیز گرفتار می شوند.ممکن است بیماری با درد وتورم مفاصل زانو ،مفاصل ران ها، شانه ها ومچ دست ها وپاها شروع شود ویا ممکن است علایم کمری ،لگن ومفاصل محیطی با هم دیده شوند.از علایم دیگری که در عده ا از بیماران دیده می شود،گرفتاری چشمی به صورت سرخی وتاری است که درنتیجه التهاب اطاق های چشمی اتفاق می افتد و اگر درمان زودرس صورت نگیرد،منجر به تاری دایمی چشم می گردد.همچنین در این بیماری ندرتا ممکن است قلب،ریتین وسایر دستگاه ها گرفتار شوند.این بیماری به ندرت می تواند ثانوی، به بیماری های دیگر مثل پسوریازیس ویا بیماری های التهابی روده ویا عفونی باشد،لذا علاوه بر علایم فوق،علایم بیماری اولیه نیز ممکن است وجود داشته باشد.در مورد درمان این بیماری که سیر مزمن نیز وجود دارد وممکن است در یک بیمار،سال ها فعال باشد،لازم است بیماران ،آگاهی کافی درمورد بیماری خود داشته باشند، لذا متخصص روماتولوژی علاوه بر درمان دارویی وسایر اقدامات دارویی لازم،باید اطلاعات کافی در مورد این بیماری در اختیار مبتلایان بگذارد.
مشخص ترین علامت این بیماری،درد ستون فقرات ولگن است و از آنجا که درد کمری، یکی از شایع ترین شکایات افراد هر جامعه است، تعداد زیادی از کسانی که مبتلا به این بیماری می شوند، ممکن است با تشخیص های دیگری درمان شوند.بسیاری از بیماران هستند که ماه ها یا سال ها قبل از اینکه به طور قطعی، تشخیص بیماری یشان داده شود، در بخش های مختلف داخل کشور، سرگردان بوده وبرای تعداد دیگری از بیماران نیز بررسی ها،تشخیص ودرمان اشتباه صورت میگیرد که این، خود عاملی است برای اضافه شدن اختلالات دیگری در ستون فقرات که درنهایت، درد ناشی از اقدامات صورت گرفته، به درد کمری و لگن اضافه می شودومشکل بیماری را برحسب وسعت اقداماتی که انجام گرفته، بیشتر می کند.درد ناشی از AS اکثرابه آهستگی و خود به خود شروع می شود و ماه ها به طور ثابت یا پیش رونده باقی می ماند.این درد با استراحت، بیشتر می شود وبا فعالیت کاهش می یابد.درد لگن وستون فقرات پشتی-کمری،اکثرا بیمار رادر نیمه دوم شب از خواب بیدار شده وبرحسب شدت درد ، مدتی راه رفته و دوباره به رختخواب برگردد.وقتی صبحگاهان فرا می رسد وبیمار،از خواب بلند می شود،درد بیشتر است وخشکی فقرات کمری ولگن وجود دارد.هرچه بیماری شدیدتر باشد، خشکی صبحگاهی بیشتر طول میکشد وگاهی در بعضی از بیماران تا نیمه روز، خشکی باقی می ماند.وقتی بیمار از رختخواب بیرون می آید،خشکی صبحگاهی و دردشدید وجود دارد ولی با شروع فعالیت، درد وخشکی، به تدریج از بین رفته واکثرا،بیمار، آن روزها احساس راحتی می کند؛در عده کمی از مبتلایان به طور کلی درد وجودندارد.یکی از خصوصیات این بیماری،التهاب محل چسبیدن رباط ها به استخوان است، لذا هر جایی که رباط ها به استخوان می چسبند،بیماران، احساس درد می کنند، مثلا درد در پاشنه ها ودرد قفسه صدری وجود داردویا حتی ممکن است در بعضی ها، شروع بیماری با درد پاشنه ها وقفسه صدری باشد.علاوه برگرفتاری مفاصل لگن،ستون فقرات،پاشنه هاوقفسه صدری ومحل چسبیدن رباط ها به استخوان ها،در این بیماری،مفاصل بزرگ اندام ها نیز گرفتار می شوند.ممکن است بیماری با درد وتورم مفاصل زانو ،مفاصل ران ها، شانه ها ومچ دست ها وپاها شروع شود ویا ممکن است علایم کمری ،لگن ومفاصل محیطی با هم دیده شوند.از علایم دیگری که در عده ا از بیماران دیده می شود،گرفتاری چشمی به صورت سرخی وتاری است که درنتیجه التهاب اطاق های چشمی اتفاق می افتد و اگر درمان زودرس صورت نگیرد،منجر به تاری دایمی چشم می گردد.همچنین در این بیماری ندرتا ممکن است قلب،ریتین وسایر دستگاه ها گرفتار شوند.این بیماری به ندرت می تواند ثانوی، به بیماری های دیگر مثل پسوریازیس ویا بیماری های التهابی روده ویا عفونی باشد،لذا علاوه بر علایم فوق،علایم بیماری اولیه نیز ممکن است وجود داشته باشد.در مورد درمان این بیماری که سیر مزمن نیز وجود دارد وممکن است در یک بیمار،سال ها فعال باشد،لازم است بیماران ،آگاهی کافی درمورد بیماری خود داشته باشند، لذا متخصص روماتولوژی علاوه بر درمان دارویی وسایر اقدامات دارویی لازم،باید اطلاعات کافی در مورد این بیماری در اختیار مبتلایان بگذارد.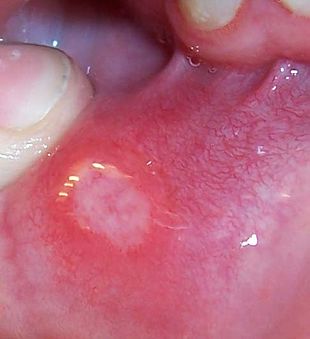
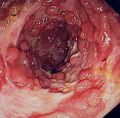 شیوع و همه گیری شناسیطبق یافته های موسسه کولیت و کرون آمریکا( CCFA)، چنین برآورد شده است که حدود نیم میلیون نفر از بیماری کرون رنج می برند. به نظر می رسد زنان و مردان به میزان مساوی مبتلا شده اند.بیماری کرون در درجه اول بر افراد بین سنین 15 و 35 سال تاثیر می گذارد ،اما می تواند کودکان و افراد مسن را به خوبی تحت تاثیر قرار دهد .CCFA تخمین می زند که 000،100 نفر را افراد کمتر از 18 سال تشکیل میدهند .CCFA همچنین می گوید که این خطر توسعه بیماری در افرادی که یکی از بستگان مبتلا هستند10 برابر است و این خطر برای افرادی که یکی از خواهر ها یا برادرها مبتلا هستند تا 30 برابر بالا میرود .همچنین یهودی های اروپایی تبار 4 تا 5 برابر بیشتر مبتلا میشوند میزان شیوع در سفید پوستان 149 در 100000میباشد و شیوع پایین در میان آسیایی ها است.علت بیماری عوامل مؤثر در بروز این بیماری شامل ارث، عوامل محیطی، اختلالات سیستم ایمنی ،باکتری های روده و نوعی از رژیم غذایی(دریافت بالای پروتئین حیوانی و دریافت پایین امگا 3 )است. علت بیماری کرون ناشناخته است. اطلاعات موجود درباره این بیماری نشان دهنده آن است که بیماری در بین اقوام و گروههای خاص بیشتر از گروه های دیگر دیده می شود و این دلالت بر آن دارد که فاکتورهای ژنتیک در بروز بیماری میتواند نقش داشته باشد. باور بر این است که در افرادی که از نظر ژنتیکی، حساس هستند سیستم ایمنی بدن توسط یک محرک تحریک شده و باعث التهاب در دستگاه گوارش می شود. همچنین باکتری و غذای موجود در دستگاه گوارش نیز در بروز بیماری نقش دارند. این آسیب عامل اصلی بروز علائم بیماری است. بیماری کرون اغلب ماه ها یا سال ها قبل از تشخیص بیماری وجود دارد و ممکن است در کودکانی که تأخیر رشد دارند تنها نشانه ظاهری بیماری باشد. علایم این بیماری شامل کاهش وزن قابل توجه، تب متناوب، اسهال مزمن آبکی و دردهای شکمی به خصوص در ناحیه یک چهارم راست پایینی شکم است که با اجابت مزاج تسکین نمی یابد.علایم ناشی از بیماری شامل زخم(آفت) دهان، اسهال، درد شکم ،کاهش وزن و تب است. همچنین بیمار ممکن است به علائم خارج رودهای از قبیل راشهای پوستی، درد مفاصل، التهاب چشم، و با شیوع کمتر اختلالات کبدی مبتلا شود. اگر چه بیماری کرون یک بیماری مزمن است، درمانهای دارویی و جراحی به کنترل بیماری کمک میکند تا بیمار برای مدت طولانی بدون علائم زندگی کند .این بیماری علاوه بر گرفتار کردن معده و روده، اندام های دیگر را نیز می تواند درگیر کند. ورم چشم و ورم مفاصل یا ماهیچه های دست یا پا از این دست هستند .نازکی یا پوکی استخوان می تواند از عواقب این بیماری باشد. در این بیماری پوست، خون و غدد درون ریز نیز می توانند درگیر شوند .بیماری ک مخونی همولیتیک خود ایمنی که در آن سیستم ایمنی به گویچه های سرخ حمله می کند بیشتر در بیماری کرون شایع است و می تواند باعث خستگی، رنگ پریدگی و دیگر نشانه های شایع در ک مخونی شود.
شیوع و همه گیری شناسیطبق یافته های موسسه کولیت و کرون آمریکا( CCFA)، چنین برآورد شده است که حدود نیم میلیون نفر از بیماری کرون رنج می برند. به نظر می رسد زنان و مردان به میزان مساوی مبتلا شده اند.بیماری کرون در درجه اول بر افراد بین سنین 15 و 35 سال تاثیر می گذارد ،اما می تواند کودکان و افراد مسن را به خوبی تحت تاثیر قرار دهد .CCFA تخمین می زند که 000،100 نفر را افراد کمتر از 18 سال تشکیل میدهند .CCFA همچنین می گوید که این خطر توسعه بیماری در افرادی که یکی از بستگان مبتلا هستند10 برابر است و این خطر برای افرادی که یکی از خواهر ها یا برادرها مبتلا هستند تا 30 برابر بالا میرود .همچنین یهودی های اروپایی تبار 4 تا 5 برابر بیشتر مبتلا میشوند میزان شیوع در سفید پوستان 149 در 100000میباشد و شیوع پایین در میان آسیایی ها است.علت بیماری عوامل مؤثر در بروز این بیماری شامل ارث، عوامل محیطی، اختلالات سیستم ایمنی ،باکتری های روده و نوعی از رژیم غذایی(دریافت بالای پروتئین حیوانی و دریافت پایین امگا 3 )است. علت بیماری کرون ناشناخته است. اطلاعات موجود درباره این بیماری نشان دهنده آن است که بیماری در بین اقوام و گروههای خاص بیشتر از گروه های دیگر دیده می شود و این دلالت بر آن دارد که فاکتورهای ژنتیک در بروز بیماری میتواند نقش داشته باشد. باور بر این است که در افرادی که از نظر ژنتیکی، حساس هستند سیستم ایمنی بدن توسط یک محرک تحریک شده و باعث التهاب در دستگاه گوارش می شود. همچنین باکتری و غذای موجود در دستگاه گوارش نیز در بروز بیماری نقش دارند. این آسیب عامل اصلی بروز علائم بیماری است. بیماری کرون اغلب ماه ها یا سال ها قبل از تشخیص بیماری وجود دارد و ممکن است در کودکانی که تأخیر رشد دارند تنها نشانه ظاهری بیماری باشد. علایم این بیماری شامل کاهش وزن قابل توجه، تب متناوب، اسهال مزمن آبکی و دردهای شکمی به خصوص در ناحیه یک چهارم راست پایینی شکم است که با اجابت مزاج تسکین نمی یابد.علایم ناشی از بیماری شامل زخم(آفت) دهان، اسهال، درد شکم ،کاهش وزن و تب است. همچنین بیمار ممکن است به علائم خارج رودهای از قبیل راشهای پوستی، درد مفاصل، التهاب چشم، و با شیوع کمتر اختلالات کبدی مبتلا شود. اگر چه بیماری کرون یک بیماری مزمن است، درمانهای دارویی و جراحی به کنترل بیماری کمک میکند تا بیمار برای مدت طولانی بدون علائم زندگی کند .این بیماری علاوه بر گرفتار کردن معده و روده، اندام های دیگر را نیز می تواند درگیر کند. ورم چشم و ورم مفاصل یا ماهیچه های دست یا پا از این دست هستند .نازکی یا پوکی استخوان می تواند از عواقب این بیماری باشد. در این بیماری پوست، خون و غدد درون ریز نیز می توانند درگیر شوند .بیماری ک مخونی همولیتیک خود ایمنی که در آن سیستم ایمنی به گویچه های سرخ حمله می کند بیشتر در بیماری کرون شایع است و می تواند باعث خستگی، رنگ پریدگی و دیگر نشانه های شایع در ک مخونی شود.
 شیوع و همه گیری شناسی بیشتر در بالغین جوان 30-40 ساله رخ می دهد. در زنان بیشتراز مردان وجود دارد. شرایط پیشرونده منجر به نارسایی قلب راست و دیس پنه شدید می شود. فشار خون ریوی اولیه جزء بیماریهای نادر بوده و میزان بروز آن 2 تا 3 در یک میلیون نفر تخمین زده می شود. این بیماری در کودکان تظاهر پیدا کرده و در زنان سه برابر مردان مشاهده میشود. از کل بیماران افزایش فشار ریوی 95-90درصد ثانویه و 10-5درصد اولیه میباشد .علت بیماری علت فشار خون اولیه نامشخص میباشد. فشارخون ریوی ثانویه زمانی است که فشارخون ریوی در اثر بیماری دیگری ایجاد شود که علل شایع آن ،مشکلات تنفسی مانندبی ماریهای انسدادی مزمن ریوی (آمفیزم و برونشیت مزمن) هستند. علل دیگر شامل نارسایی احتقانی قلب، نقص مادرزادی قلب، وجود لخته در عروق ریوی، ایدز(سندرم نقص ایمنی مادرزادی)، سیروز(یک نوع بیماری کبدی مزمن)، لوپوس ،فیبروز ریوی (شرایطی که باعث ایجاد اسکار در ریه می شود) و داروهایی خاص است. هر دو نوع فشارخون ریوی(اولیه و ثانویه) معمولاً دائمی هستند و درمان تنها باعث کاهش علایم و بهبود زندگی بیمار میشود.علایم ناشی از بیماری بیماران با مشکل خفیف معمولا بدون علامت هستند. در فرم متوسط تا شدید علامت اصلی و معمولا تنها علامت تنگی نفس است. تنگی نفس ابتدا هنگام فعالیت و بعدا در زمان استراحت نیز ظاهر می شود. درد زیر جناغ شایع است که در 50 – 25% بیماران دیده می شود. سایر علائم شامل خستگی، سنکوب، درد قفسه سینه مشابه آنژین، تپش قلب، ضعف عضلانی، سرفه غیر موثر، علائم نارسایی طرف راست قلب شامل ادم محیطی ،آسیت ،اتساع وریدهای گردنی، احتقان کبد، رال و سوفل قلبی، قاشقی شدن انگشتان میباشند .هنگامیکه بیماری پیشرفت کرد، علایم بدتر می شوند و می تواند شامل علایم زیر باشد:
شیوع و همه گیری شناسی بیشتر در بالغین جوان 30-40 ساله رخ می دهد. در زنان بیشتراز مردان وجود دارد. شرایط پیشرونده منجر به نارسایی قلب راست و دیس پنه شدید می شود. فشار خون ریوی اولیه جزء بیماریهای نادر بوده و میزان بروز آن 2 تا 3 در یک میلیون نفر تخمین زده می شود. این بیماری در کودکان تظاهر پیدا کرده و در زنان سه برابر مردان مشاهده میشود. از کل بیماران افزایش فشار ریوی 95-90درصد ثانویه و 10-5درصد اولیه میباشد .علت بیماری علت فشار خون اولیه نامشخص میباشد. فشارخون ریوی ثانویه زمانی است که فشارخون ریوی در اثر بیماری دیگری ایجاد شود که علل شایع آن ،مشکلات تنفسی مانندبی ماریهای انسدادی مزمن ریوی (آمفیزم و برونشیت مزمن) هستند. علل دیگر شامل نارسایی احتقانی قلب، نقص مادرزادی قلب، وجود لخته در عروق ریوی، ایدز(سندرم نقص ایمنی مادرزادی)، سیروز(یک نوع بیماری کبدی مزمن)، لوپوس ،فیبروز ریوی (شرایطی که باعث ایجاد اسکار در ریه می شود) و داروهایی خاص است. هر دو نوع فشارخون ریوی(اولیه و ثانویه) معمولاً دائمی هستند و درمان تنها باعث کاهش علایم و بهبود زندگی بیمار میشود.علایم ناشی از بیماری بیماران با مشکل خفیف معمولا بدون علامت هستند. در فرم متوسط تا شدید علامت اصلی و معمولا تنها علامت تنگی نفس است. تنگی نفس ابتدا هنگام فعالیت و بعدا در زمان استراحت نیز ظاهر می شود. درد زیر جناغ شایع است که در 50 – 25% بیماران دیده می شود. سایر علائم شامل خستگی، سنکوب، درد قفسه سینه مشابه آنژین، تپش قلب، ضعف عضلانی، سرفه غیر موثر، علائم نارسایی طرف راست قلب شامل ادم محیطی ،آسیت ،اتساع وریدهای گردنی، احتقان کبد، رال و سوفل قلبی، قاشقی شدن انگشتان میباشند .هنگامیکه بیماری پیشرفت کرد، علایم بدتر می شوند و می تواند شامل علایم زیر باشد: