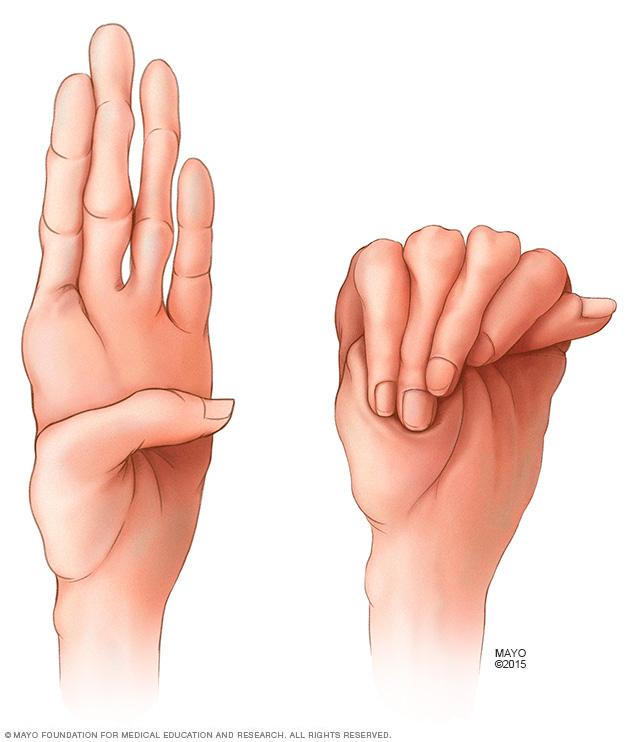
 شیوع و همه گیری شناسی این بیماری معمولا در هر 10000 نفر یک مورد ممکن است وجود داشته باشد ولی گفته شده ممکن است بین 5000-3000 هم وجود داشته باشد. هم اکنون در آمریکا 200 هزار بیمار دچار سندرم مارفان وجود دارد.هم در کودکان و هم در بالغین و در زن و مرد ممکن است رخ دهد.این اختلال از ابتدای تولد وجود داشته و گاهی در نوزادان قابل تشخیص است . با این حال ، علایم آن گاهی تا نوجوانی یا جوانی ظاهر نشده و شدت علایم نیز بسیار متغیر است. شیوع آن در خانم ها و آقایان برابر است .علت بیماری این ناهنجاری به علت جهش در کروموزوم 15رخ میدهد. تکه 1FBN، بخشی از کروموزوم 15 است که دچار اختلال میگردد .وظیفه این تکه بیان دستورالعمل ساخت فیبریلین است. زمانی که این ژن به درستی بیان نشود دستور ساخت فیبریلین به درستی بیان نشده و پروتئین ساختار اصلی خود را نخواهد داشت. این پروتئین نقش مهمی در ساختار بافتهای پیوندی دارد. سراسر بدن از بافتهای پیوندی تشکیل شده است. بنابر این ابتلا به این سندرم ،بسیاری از ارگانها را درگیر میکند مانند دستگاه اسکلتی، چشم ،قلب، آئورت، عروق خونی، سیستم عصبی، پوست و ریهعلایم ناشی از بیماری علایم استخوانی عضلانی :قامت بلند و بدن لاغر و کشی ده طول اندامها نسبت به تنه نامتناسب است.انگشتان باریک و بلند( انگشتان عنکبوتی)شکل غیرطبیعی قفسه سینه بلندی قوس کاممفصل دوگانه؛ ضعف یا نرمی مفصلعلایم قلبی عروقی : نارسایی دریچه آئورت ؛ شکافت آئورت پرولاپس یا نارسایی دریچه میترالجابجایی عدسی چشم ، معمولاً به سمت بالا نزدیک بینی جداشدگی شبکیه (ناشایع )گلوکوم (آب سیاه) و یا کاتاراکت ( آب مروارید)سایر علایم : کبود شدن آسان پوست (ناشایع )خونریزی بیش از حد معمول (ناشایع)روش تشخیص هیچ آزمایش تشخیصی برای سندرم مارفان وجود ندارد. تشخیص پزشک تنها بواسطه مشاهده و تاریخچه کامل پزشکی است. اطلاعات کامل درباره همه اعضاء خانواده که ممکن است مشکل و یا مرگ زود هنگام مرتبط با بیماریهای قلبی داشته اند. ارزیابی بالینی کامل شامل: ارزیابی سیستم اسکلتی، نسبت اندازه بازو و پا به قد و آزمایشات چشمی. تستهای قلبی از قبیل اکوگاردیوگرام برای سنجش قلب و آئورت.درمان متاسفانه سندرم مارفان همانند سایر بیماریهای ژنتیکی قابل درمان نیست و هیچ راهی برای اصلاح ناهنجاری بافت پیوندی وجود ندارد. هدف درمان ثابت نگه داشتن روند بیماری قبل از اینکه به مراحل حاد رسیده و عوارض خطرناکی را ایجاد نماید. برخی از افراد دارو تجویز می نمایند که استفاده از این داروها نیز خود یک بحث چالش انگیز است. بتابلوکرها فرآیند دیلاتاسیون را به تاخیر می اندازند. آنتی کوآگولانت از قبیل وارفارین پس از جایگزینی دریچه قلب مصنوعی ضرورت می یابد. آنتی بیوتیک تراپی داخل وریدی در طی روشهای قلبی برای جلوگیری از آندوکاردیت باکتریال ضروری است .جراحی های قلبی عروقی امکان بقا بیشتر بیمار را فراهم می آورد. در برخی موارد اسکلیوز شدید نیز نیاز به مداخله شدید جراحی دارد. استفاده از لیزر نیز برای درمان کندگی شبکیه مفید است.مراقبت های لازم در حال حاضر هیچ روش تشخیصی قبل از تولد در مورد این بیماری وجود ندارد.در صورت مبتلا بودن یکی از والدین ، احتمال درگیری هریک از فرزندان 50% خواهد بود. البته با توجه به متغیر بودن شدت علایم این بیماری در بیماران مختلف، شدت علایم در فرزندان مبتلا ممکن است بیشتر یا کمتر از والدین باشد. در صورتی که دچار این بیماری بوده یا سابقه خانوادگی آن را دارید قبل از ازدواج ؛ مشاوره ژنتیک را حتماً مدنظر داشته باشید .تا آنجا که علایم بیماری به شما اجازه می دهد فعال باشید .افراد دچار این اختلال به علت احتمال خطر بروز مرگ ناگهانی باید از شرکت در ورزش های هوازی خودداری کنند.رژیم غذایی: رژیم خاصی نیاز نیست .پیش آگهی عوارض قلبی عروقی این بیماری می تواند تهدیدکننده حیات باشد. قبل از پیدایش جراحی اصلاحی بیشتر بیماران دچار این بی ماری تا قبل از سن 35 سالگی فوت می کردند. با مداخله جراحی، بیشتر بیماران طول عمر طبی عی خواهند داشت.سندرم مارفان یک اختلال مادام العمر است .پیش آگهی بیماری در س الهای اخیر بیشتر شده است. تشخیص زودرس و پیشرفت تکنولوژی پزشکی کیفیت زندگی بیماران را بهبود بخشیده است. در سال های قبل افراد مبتلا به سندرم مارفان در سن چهل سالگی میمردند اما امروزه بیشتر افراد مبتلا تا شصت سالگی عمر می کنند. شاید پیشگیری از اتساع بیش از حد آئورت و گسیختگی آن توجیهی برای افزایش طول عمر بیماران باشد .
شیوع و همه گیری شناسی این بیماری معمولا در هر 10000 نفر یک مورد ممکن است وجود داشته باشد ولی گفته شده ممکن است بین 5000-3000 هم وجود داشته باشد. هم اکنون در آمریکا 200 هزار بیمار دچار سندرم مارفان وجود دارد.هم در کودکان و هم در بالغین و در زن و مرد ممکن است رخ دهد.این اختلال از ابتدای تولد وجود داشته و گاهی در نوزادان قابل تشخیص است . با این حال ، علایم آن گاهی تا نوجوانی یا جوانی ظاهر نشده و شدت علایم نیز بسیار متغیر است. شیوع آن در خانم ها و آقایان برابر است .علت بیماری این ناهنجاری به علت جهش در کروموزوم 15رخ میدهد. تکه 1FBN، بخشی از کروموزوم 15 است که دچار اختلال میگردد .وظیفه این تکه بیان دستورالعمل ساخت فیبریلین است. زمانی که این ژن به درستی بیان نشود دستور ساخت فیبریلین به درستی بیان نشده و پروتئین ساختار اصلی خود را نخواهد داشت. این پروتئین نقش مهمی در ساختار بافتهای پیوندی دارد. سراسر بدن از بافتهای پیوندی تشکیل شده است. بنابر این ابتلا به این سندرم ،بسیاری از ارگانها را درگیر میکند مانند دستگاه اسکلتی، چشم ،قلب، آئورت، عروق خونی، سیستم عصبی، پوست و ریهعلایم ناشی از بیماری علایم استخوانی عضلانی :قامت بلند و بدن لاغر و کشی ده طول اندامها نسبت به تنه نامتناسب است.انگشتان باریک و بلند( انگشتان عنکبوتی)شکل غیرطبیعی قفسه سینه بلندی قوس کاممفصل دوگانه؛ ضعف یا نرمی مفصلعلایم قلبی عروقی : نارسایی دریچه آئورت ؛ شکافت آئورت پرولاپس یا نارسایی دریچه میترالجابجایی عدسی چشم ، معمولاً به سمت بالا نزدیک بینی جداشدگی شبکیه (ناشایع )گلوکوم (آب سیاه) و یا کاتاراکت ( آب مروارید)سایر علایم : کبود شدن آسان پوست (ناشایع )خونریزی بیش از حد معمول (ناشایع)روش تشخیص هیچ آزمایش تشخیصی برای سندرم مارفان وجود ندارد. تشخیص پزشک تنها بواسطه مشاهده و تاریخچه کامل پزشکی است. اطلاعات کامل درباره همه اعضاء خانواده که ممکن است مشکل و یا مرگ زود هنگام مرتبط با بیماریهای قلبی داشته اند. ارزیابی بالینی کامل شامل: ارزیابی سیستم اسکلتی، نسبت اندازه بازو و پا به قد و آزمایشات چشمی. تستهای قلبی از قبیل اکوگاردیوگرام برای سنجش قلب و آئورت.درمان متاسفانه سندرم مارفان همانند سایر بیماریهای ژنتیکی قابل درمان نیست و هیچ راهی برای اصلاح ناهنجاری بافت پیوندی وجود ندارد. هدف درمان ثابت نگه داشتن روند بیماری قبل از اینکه به مراحل حاد رسیده و عوارض خطرناکی را ایجاد نماید. برخی از افراد دارو تجویز می نمایند که استفاده از این داروها نیز خود یک بحث چالش انگیز است. بتابلوکرها فرآیند دیلاتاسیون را به تاخیر می اندازند. آنتی کوآگولانت از قبیل وارفارین پس از جایگزینی دریچه قلب مصنوعی ضرورت می یابد. آنتی بیوتیک تراپی داخل وریدی در طی روشهای قلبی برای جلوگیری از آندوکاردیت باکتریال ضروری است .جراحی های قلبی عروقی امکان بقا بیشتر بیمار را فراهم می آورد. در برخی موارد اسکلیوز شدید نیز نیاز به مداخله شدید جراحی دارد. استفاده از لیزر نیز برای درمان کندگی شبکیه مفید است.مراقبت های لازم در حال حاضر هیچ روش تشخیصی قبل از تولد در مورد این بیماری وجود ندارد.در صورت مبتلا بودن یکی از والدین ، احتمال درگیری هریک از فرزندان 50% خواهد بود. البته با توجه به متغیر بودن شدت علایم این بیماری در بیماران مختلف، شدت علایم در فرزندان مبتلا ممکن است بیشتر یا کمتر از والدین باشد. در صورتی که دچار این بیماری بوده یا سابقه خانوادگی آن را دارید قبل از ازدواج ؛ مشاوره ژنتیک را حتماً مدنظر داشته باشید .تا آنجا که علایم بیماری به شما اجازه می دهد فعال باشید .افراد دچار این اختلال به علت احتمال خطر بروز مرگ ناگهانی باید از شرکت در ورزش های هوازی خودداری کنند.رژیم غذایی: رژیم خاصی نیاز نیست .پیش آگهی عوارض قلبی عروقی این بیماری می تواند تهدیدکننده حیات باشد. قبل از پیدایش جراحی اصلاحی بیشتر بیماران دچار این بی ماری تا قبل از سن 35 سالگی فوت می کردند. با مداخله جراحی، بیشتر بیماران طول عمر طبی عی خواهند داشت.سندرم مارفان یک اختلال مادام العمر است .پیش آگهی بیماری در س الهای اخیر بیشتر شده است. تشخیص زودرس و پیشرفت تکنولوژی پزشکی کیفیت زندگی بیماران را بهبود بخشیده است. در سال های قبل افراد مبتلا به سندرم مارفان در سن چهل سالگی میمردند اما امروزه بیشتر افراد مبتلا تا شصت سالگی عمر می کنند. شاید پیشگیری از اتساع بیش از حد آئورت و گسیختگی آن توجیهی برای افزایش طول عمر بیماران باشد . 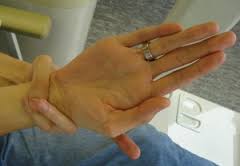
۱۵ آبان۱۳۹۵
سندرم مارفان (Marfan Syndrome)
سندرم مارفان یک اختلال ژنتیکی میباشد. این ناهنجاری به علت جهش در کروموزوم 15رخ میدهد.وظیفه این ژن بیان دستورالعمل ساخت فیبریلین است. سندرم مارفان نوعی اختلال وراثتی است که در اصل بافت همبندی بدن را هدف قرار میدهد و بیماران مبتلا به این سندرم دارای استخوان های بلندتر و عضلاتی شل تر از حد طبیعی هستند. در چشم ها و دستگاه قلب و عروق آنان اختلالات زیادی به وجود می آید . شیوع و همه گیری شناسی این بیماری معمولا در هر 10000 نفر یک مورد ممکن است وجود داشته باشد ولی گفته شده ممکن است بین 5000-3000 هم وجود داشته باشد. هم اکنون در آمریکا 200 هزار بیمار دچار سندرم مارفان وجود دارد.هم در کودکان و هم در بالغین و در زن و مرد ممکن است رخ دهد.این اختلال از ابتدای تولد وجود داشته و گاهی در نوزادان قابل تشخیص است . با این حال ، علایم آن گاهی تا نوجوانی یا جوانی ظاهر نشده و شدت علایم نیز بسیار متغیر است. شیوع آن در خانم ها و آقایان برابر است .علت بیماری این ناهنجاری به علت جهش در کروموزوم 15رخ میدهد. تکه 1FBN، بخشی از کروموزوم 15 است که دچار اختلال میگردد .وظیفه این تکه بیان دستورالعمل ساخت فیبریلین است. زمانی که این ژن به درستی بیان نشود دستور ساخت فیبریلین به درستی بیان نشده و پروتئین ساختار اصلی خود را نخواهد داشت. این پروتئین نقش مهمی در ساختار بافتهای پیوندی دارد. سراسر بدن از بافتهای پیوندی تشکیل شده است. بنابر این ابتلا به این سندرم ،بسیاری از ارگانها را درگیر میکند مانند دستگاه اسکلتی، چشم ،قلب، آئورت، عروق خونی، سیستم عصبی، پوست و ریهعلایم ناشی از بیماری علایم استخوانی عضلانی :قامت بلند و بدن لاغر و کشی ده طول اندامها نسبت به تنه نامتناسب است.انگشتان باریک و بلند( انگشتان عنکبوتی)شکل غیرطبیعی قفسه سینه بلندی قوس کاممفصل دوگانه؛ ضعف یا نرمی مفصلعلایم قلبی عروقی : نارسایی دریچه آئورت ؛ شکافت آئورت پرولاپس یا نارسایی دریچه میترالجابجایی عدسی چشم ، معمولاً به سمت بالا نزدیک بینی جداشدگی شبکیه (ناشایع )گلوکوم (آب سیاه) و یا کاتاراکت ( آب مروارید)سایر علایم : کبود شدن آسان پوست (ناشایع )خونریزی بیش از حد معمول (ناشایع)روش تشخیص هیچ آزمایش تشخیصی برای سندرم مارفان وجود ندارد. تشخیص پزشک تنها بواسطه مشاهده و تاریخچه کامل پزشکی است. اطلاعات کامل درباره همه اعضاء خانواده که ممکن است مشکل و یا مرگ زود هنگام مرتبط با بیماریهای قلبی داشته اند. ارزیابی بالینی کامل شامل: ارزیابی سیستم اسکلتی، نسبت اندازه بازو و پا به قد و آزمایشات چشمی. تستهای قلبی از قبیل اکوگاردیوگرام برای سنجش قلب و آئورت.درمان متاسفانه سندرم مارفان همانند سایر بیماریهای ژنتیکی قابل درمان نیست و هیچ راهی برای اصلاح ناهنجاری بافت پیوندی وجود ندارد. هدف درمان ثابت نگه داشتن روند بیماری قبل از اینکه به مراحل حاد رسیده و عوارض خطرناکی را ایجاد نماید. برخی از افراد دارو تجویز می نمایند که استفاده از این داروها نیز خود یک بحث چالش انگیز است. بتابلوکرها فرآیند دیلاتاسیون را به تاخیر می اندازند. آنتی کوآگولانت از قبیل وارفارین پس از جایگزینی دریچه قلب مصنوعی ضرورت می یابد. آنتی بیوتیک تراپی داخل وریدی در طی روشهای قلبی برای جلوگیری از آندوکاردیت باکتریال ضروری است .جراحی های قلبی عروقی امکان بقا بیشتر بیمار را فراهم می آورد. در برخی موارد اسکلیوز شدید نیز نیاز به مداخله شدید جراحی دارد. استفاده از لیزر نیز برای درمان کندگی شبکیه مفید است.مراقبت های لازم در حال حاضر هیچ روش تشخیصی قبل از تولد در مورد این بیماری وجود ندارد.در صورت مبتلا بودن یکی از والدین ، احتمال درگیری هریک از فرزندان 50% خواهد بود. البته با توجه به متغیر بودن شدت علایم این بیماری در بیماران مختلف، شدت علایم در فرزندان مبتلا ممکن است بیشتر یا کمتر از والدین باشد. در صورتی که دچار این بیماری بوده یا سابقه خانوادگی آن را دارید قبل از ازدواج ؛ مشاوره ژنتیک را حتماً مدنظر داشته باشید .تا آنجا که علایم بیماری به شما اجازه می دهد فعال باشید .افراد دچار این اختلال به علت احتمال خطر بروز مرگ ناگهانی باید از شرکت در ورزش های هوازی خودداری کنند.رژیم غذایی: رژیم خاصی نیاز نیست .پیش آگهی عوارض قلبی عروقی این بیماری می تواند تهدیدکننده حیات باشد. قبل از پیدایش جراحی اصلاحی بیشتر بیماران دچار این بی ماری تا قبل از سن 35 سالگی فوت می کردند. با مداخله جراحی، بیشتر بیماران طول عمر طبی عی خواهند داشت.سندرم مارفان یک اختلال مادام العمر است .پیش آگهی بیماری در س الهای اخیر بیشتر شده است. تشخیص زودرس و پیشرفت تکنولوژی پزشکی کیفیت زندگی بیماران را بهبود بخشیده است. در سال های قبل افراد مبتلا به سندرم مارفان در سن چهل سالگی میمردند اما امروزه بیشتر افراد مبتلا تا شصت سالگی عمر می کنند. شاید پیشگیری از اتساع بیش از حد آئورت و گسیختگی آن توجیهی برای افزایش طول عمر بیماران باشد .
شیوع و همه گیری شناسی این بیماری معمولا در هر 10000 نفر یک مورد ممکن است وجود داشته باشد ولی گفته شده ممکن است بین 5000-3000 هم وجود داشته باشد. هم اکنون در آمریکا 200 هزار بیمار دچار سندرم مارفان وجود دارد.هم در کودکان و هم در بالغین و در زن و مرد ممکن است رخ دهد.این اختلال از ابتدای تولد وجود داشته و گاهی در نوزادان قابل تشخیص است . با این حال ، علایم آن گاهی تا نوجوانی یا جوانی ظاهر نشده و شدت علایم نیز بسیار متغیر است. شیوع آن در خانم ها و آقایان برابر است .علت بیماری این ناهنجاری به علت جهش در کروموزوم 15رخ میدهد. تکه 1FBN، بخشی از کروموزوم 15 است که دچار اختلال میگردد .وظیفه این تکه بیان دستورالعمل ساخت فیبریلین است. زمانی که این ژن به درستی بیان نشود دستور ساخت فیبریلین به درستی بیان نشده و پروتئین ساختار اصلی خود را نخواهد داشت. این پروتئین نقش مهمی در ساختار بافتهای پیوندی دارد. سراسر بدن از بافتهای پیوندی تشکیل شده است. بنابر این ابتلا به این سندرم ،بسیاری از ارگانها را درگیر میکند مانند دستگاه اسکلتی، چشم ،قلب، آئورت، عروق خونی، سیستم عصبی، پوست و ریهعلایم ناشی از بیماری علایم استخوانی عضلانی :قامت بلند و بدن لاغر و کشی ده طول اندامها نسبت به تنه نامتناسب است.انگشتان باریک و بلند( انگشتان عنکبوتی)شکل غیرطبیعی قفسه سینه بلندی قوس کاممفصل دوگانه؛ ضعف یا نرمی مفصلعلایم قلبی عروقی : نارسایی دریچه آئورت ؛ شکافت آئورت پرولاپس یا نارسایی دریچه میترالجابجایی عدسی چشم ، معمولاً به سمت بالا نزدیک بینی جداشدگی شبکیه (ناشایع )گلوکوم (آب سیاه) و یا کاتاراکت ( آب مروارید)سایر علایم : کبود شدن آسان پوست (ناشایع )خونریزی بیش از حد معمول (ناشایع)روش تشخیص هیچ آزمایش تشخیصی برای سندرم مارفان وجود ندارد. تشخیص پزشک تنها بواسطه مشاهده و تاریخچه کامل پزشکی است. اطلاعات کامل درباره همه اعضاء خانواده که ممکن است مشکل و یا مرگ زود هنگام مرتبط با بیماریهای قلبی داشته اند. ارزیابی بالینی کامل شامل: ارزیابی سیستم اسکلتی، نسبت اندازه بازو و پا به قد و آزمایشات چشمی. تستهای قلبی از قبیل اکوگاردیوگرام برای سنجش قلب و آئورت.درمان متاسفانه سندرم مارفان همانند سایر بیماریهای ژنتیکی قابل درمان نیست و هیچ راهی برای اصلاح ناهنجاری بافت پیوندی وجود ندارد. هدف درمان ثابت نگه داشتن روند بیماری قبل از اینکه به مراحل حاد رسیده و عوارض خطرناکی را ایجاد نماید. برخی از افراد دارو تجویز می نمایند که استفاده از این داروها نیز خود یک بحث چالش انگیز است. بتابلوکرها فرآیند دیلاتاسیون را به تاخیر می اندازند. آنتی کوآگولانت از قبیل وارفارین پس از جایگزینی دریچه قلب مصنوعی ضرورت می یابد. آنتی بیوتیک تراپی داخل وریدی در طی روشهای قلبی برای جلوگیری از آندوکاردیت باکتریال ضروری است .جراحی های قلبی عروقی امکان بقا بیشتر بیمار را فراهم می آورد. در برخی موارد اسکلیوز شدید نیز نیاز به مداخله شدید جراحی دارد. استفاده از لیزر نیز برای درمان کندگی شبکیه مفید است.مراقبت های لازم در حال حاضر هیچ روش تشخیصی قبل از تولد در مورد این بیماری وجود ندارد.در صورت مبتلا بودن یکی از والدین ، احتمال درگیری هریک از فرزندان 50% خواهد بود. البته با توجه به متغیر بودن شدت علایم این بیماری در بیماران مختلف، شدت علایم در فرزندان مبتلا ممکن است بیشتر یا کمتر از والدین باشد. در صورتی که دچار این بیماری بوده یا سابقه خانوادگی آن را دارید قبل از ازدواج ؛ مشاوره ژنتیک را حتماً مدنظر داشته باشید .تا آنجا که علایم بیماری به شما اجازه می دهد فعال باشید .افراد دچار این اختلال به علت احتمال خطر بروز مرگ ناگهانی باید از شرکت در ورزش های هوازی خودداری کنند.رژیم غذایی: رژیم خاصی نیاز نیست .پیش آگهی عوارض قلبی عروقی این بیماری می تواند تهدیدکننده حیات باشد. قبل از پیدایش جراحی اصلاحی بیشتر بیماران دچار این بی ماری تا قبل از سن 35 سالگی فوت می کردند. با مداخله جراحی، بیشتر بیماران طول عمر طبی عی خواهند داشت.سندرم مارفان یک اختلال مادام العمر است .پیش آگهی بیماری در س الهای اخیر بیشتر شده است. تشخیص زودرس و پیشرفت تکنولوژی پزشکی کیفیت زندگی بیماران را بهبود بخشیده است. در سال های قبل افراد مبتلا به سندرم مارفان در سن چهل سالگی میمردند اما امروزه بیشتر افراد مبتلا تا شصت سالگی عمر می کنند. شاید پیشگیری از اتساع بیش از حد آئورت و گسیختگی آن توجیهی برای افزایش طول عمر بیماران باشد .
شیوع و همه گیری شناسی این بیماری معمولا در هر 10000 نفر یک مورد ممکن است وجود داشته باشد ولی گفته شده ممکن است بین 5000-3000 هم وجود داشته باشد. هم اکنون در آمریکا 200 هزار بیمار دچار سندرم مارفان وجود دارد.هم در کودکان و هم در بالغین و در زن و مرد ممکن است رخ دهد.این اختلال از ابتدای تولد وجود داشته و گاهی در نوزادان قابل تشخیص است . با این حال ، علایم آن گاهی تا نوجوانی یا جوانی ظاهر نشده و شدت علایم نیز بسیار متغیر است. شیوع آن در خانم ها و آقایان برابر است .علت بیماری این ناهنجاری به علت جهش در کروموزوم 15رخ میدهد. تکه 1FBN، بخشی از کروموزوم 15 است که دچار اختلال میگردد .وظیفه این تکه بیان دستورالعمل ساخت فیبریلین است. زمانی که این ژن به درستی بیان نشود دستور ساخت فیبریلین به درستی بیان نشده و پروتئین ساختار اصلی خود را نخواهد داشت. این پروتئین نقش مهمی در ساختار بافتهای پیوندی دارد. سراسر بدن از بافتهای پیوندی تشکیل شده است. بنابر این ابتلا به این سندرم ،بسیاری از ارگانها را درگیر میکند مانند دستگاه اسکلتی، چشم ،قلب، آئورت، عروق خونی، سیستم عصبی، پوست و ریهعلایم ناشی از بیماری علایم استخوانی عضلانی :قامت بلند و بدن لاغر و کشی ده طول اندامها نسبت به تنه نامتناسب است.انگشتان باریک و بلند( انگشتان عنکبوتی)شکل غیرطبیعی قفسه سینه بلندی قوس کاممفصل دوگانه؛ ضعف یا نرمی مفصلعلایم قلبی عروقی : نارسایی دریچه آئورت ؛ شکافت آئورت پرولاپس یا نارسایی دریچه میترالجابجایی عدسی چشم ، معمولاً به سمت بالا نزدیک بینی جداشدگی شبکیه (ناشایع )گلوکوم (آب سیاه) و یا کاتاراکت ( آب مروارید)سایر علایم : کبود شدن آسان پوست (ناشایع )خونریزی بیش از حد معمول (ناشایع)روش تشخیص هیچ آزمایش تشخیصی برای سندرم مارفان وجود ندارد. تشخیص پزشک تنها بواسطه مشاهده و تاریخچه کامل پزشکی است. اطلاعات کامل درباره همه اعضاء خانواده که ممکن است مشکل و یا مرگ زود هنگام مرتبط با بیماریهای قلبی داشته اند. ارزیابی بالینی کامل شامل: ارزیابی سیستم اسکلتی، نسبت اندازه بازو و پا به قد و آزمایشات چشمی. تستهای قلبی از قبیل اکوگاردیوگرام برای سنجش قلب و آئورت.درمان متاسفانه سندرم مارفان همانند سایر بیماریهای ژنتیکی قابل درمان نیست و هیچ راهی برای اصلاح ناهنجاری بافت پیوندی وجود ندارد. هدف درمان ثابت نگه داشتن روند بیماری قبل از اینکه به مراحل حاد رسیده و عوارض خطرناکی را ایجاد نماید. برخی از افراد دارو تجویز می نمایند که استفاده از این داروها نیز خود یک بحث چالش انگیز است. بتابلوکرها فرآیند دیلاتاسیون را به تاخیر می اندازند. آنتی کوآگولانت از قبیل وارفارین پس از جایگزینی دریچه قلب مصنوعی ضرورت می یابد. آنتی بیوتیک تراپی داخل وریدی در طی روشهای قلبی برای جلوگیری از آندوکاردیت باکتریال ضروری است .جراحی های قلبی عروقی امکان بقا بیشتر بیمار را فراهم می آورد. در برخی موارد اسکلیوز شدید نیز نیاز به مداخله شدید جراحی دارد. استفاده از لیزر نیز برای درمان کندگی شبکیه مفید است.مراقبت های لازم در حال حاضر هیچ روش تشخیصی قبل از تولد در مورد این بیماری وجود ندارد.در صورت مبتلا بودن یکی از والدین ، احتمال درگیری هریک از فرزندان 50% خواهد بود. البته با توجه به متغیر بودن شدت علایم این بیماری در بیماران مختلف، شدت علایم در فرزندان مبتلا ممکن است بیشتر یا کمتر از والدین باشد. در صورتی که دچار این بیماری بوده یا سابقه خانوادگی آن را دارید قبل از ازدواج ؛ مشاوره ژنتیک را حتماً مدنظر داشته باشید .تا آنجا که علایم بیماری به شما اجازه می دهد فعال باشید .افراد دچار این اختلال به علت احتمال خطر بروز مرگ ناگهانی باید از شرکت در ورزش های هوازی خودداری کنند.رژیم غذایی: رژیم خاصی نیاز نیست .پیش آگهی عوارض قلبی عروقی این بیماری می تواند تهدیدکننده حیات باشد. قبل از پیدایش جراحی اصلاحی بیشتر بیماران دچار این بی ماری تا قبل از سن 35 سالگی فوت می کردند. با مداخله جراحی، بیشتر بیماران طول عمر طبی عی خواهند داشت.سندرم مارفان یک اختلال مادام العمر است .پیش آگهی بیماری در س الهای اخیر بیشتر شده است. تشخیص زودرس و پیشرفت تکنولوژی پزشکی کیفیت زندگی بیماران را بهبود بخشیده است. در سال های قبل افراد مبتلا به سندرم مارفان در سن چهل سالگی میمردند اما امروزه بیشتر افراد مبتلا تا شصت سالگی عمر می کنند. شاید پیشگیری از اتساع بیش از حد آئورت و گسیختگی آن توجیهی برای افزایش طول عمر بیماران باشد . 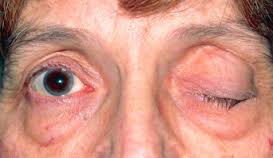
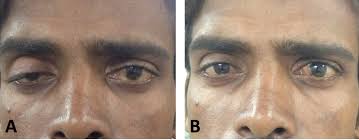 شیوع و همه گیری شناسی
این بیماری نوجوانان و بزرگسالان جوان از هر دو جنس را مبتلا می سازد ولی در خانم ها شایع تر است. بیشتر، زنان را گرفتار می کند. شیوع آن بیشتر در دهه های سوم یا چهارم زندگی است ولی امکان دارد در هر دوره ای از زندگی، از بچگی تا پیری، دیده شود. این بیماری ممکن است در هر سنی رخ دهد اما در بین خانمهای جوان در سنین 20-40سال شایع تر است ؛ اگرچه میزان شیوع بیماری در آقایان در دهه های هفتم و هشتم زندگی افزایش می یابد. به طور کلی ازهر 100هزار نفر ،14مورد ابتلا به میاستنی گراو مشاهده می شود. حداکثر وقوع آن در خانمها 20 تا 30 سالگی و در مردها دهه 6 و 7 گزارش شده است. در زیر 40 سالگی زنان 2 تا 3 برابر مردان درگیر می شوند و در اواخر عمر در مردان شایع تر می باشد .شیوع آن در برخی کشورها از 43 تا 84 در میلیون تا 14 در 100000ذکر شده است. موارد فامیلی آن نیز دیده شده است .علت بیماری علت دقیق بیماری شناخته نشده است. در بعضی موارد به همراه سایر اختلالات خودایمنی رخ میدهد و در موارد نادر بیماری در نتیجه تومورهای غده تیموس(بخشی از سیستم ایمنی محسوب می شود)ایجاد می گردد. به هر علتی که بیماری ایجاد شده باشد اختلال اصلی به دلیل حمله آنتی بادی ها (سربازان دفاعی بدن) به محل اتصال عصب به عضله رخ میدهد به این ترتیب که فعالیت عوامل شیمیایی (استیل کولین)که پیام را از عصب به عضله منتقل می کنند دچار اشکال می شود و این اختلال منجر به ضعف در عضلات و کاهش کارآیی آنها میشود.علایم ناشی از بیماریافتادگی پلک هادوبینیبه هم خوردن حالت طبیعی چهرهاختلال در بلعضعف اندامهای فوقانی و تحتانی اختلال در تکلم واضح
شیوع و همه گیری شناسی
این بیماری نوجوانان و بزرگسالان جوان از هر دو جنس را مبتلا می سازد ولی در خانم ها شایع تر است. بیشتر، زنان را گرفتار می کند. شیوع آن بیشتر در دهه های سوم یا چهارم زندگی است ولی امکان دارد در هر دوره ای از زندگی، از بچگی تا پیری، دیده شود. این بیماری ممکن است در هر سنی رخ دهد اما در بین خانمهای جوان در سنین 20-40سال شایع تر است ؛ اگرچه میزان شیوع بیماری در آقایان در دهه های هفتم و هشتم زندگی افزایش می یابد. به طور کلی ازهر 100هزار نفر ،14مورد ابتلا به میاستنی گراو مشاهده می شود. حداکثر وقوع آن در خانمها 20 تا 30 سالگی و در مردها دهه 6 و 7 گزارش شده است. در زیر 40 سالگی زنان 2 تا 3 برابر مردان درگیر می شوند و در اواخر عمر در مردان شایع تر می باشد .شیوع آن در برخی کشورها از 43 تا 84 در میلیون تا 14 در 100000ذکر شده است. موارد فامیلی آن نیز دیده شده است .علت بیماری علت دقیق بیماری شناخته نشده است. در بعضی موارد به همراه سایر اختلالات خودایمنی رخ میدهد و در موارد نادر بیماری در نتیجه تومورهای غده تیموس(بخشی از سیستم ایمنی محسوب می شود)ایجاد می گردد. به هر علتی که بیماری ایجاد شده باشد اختلال اصلی به دلیل حمله آنتی بادی ها (سربازان دفاعی بدن) به محل اتصال عصب به عضله رخ میدهد به این ترتیب که فعالیت عوامل شیمیایی (استیل کولین)که پیام را از عصب به عضله منتقل می کنند دچار اشکال می شود و این اختلال منجر به ضعف در عضلات و کاهش کارآیی آنها میشود.علایم ناشی از بیماریافتادگی پلک هادوبینیبه هم خوردن حالت طبیعی چهرهاختلال در بلعضعف اندامهای فوقانی و تحتانی اختلال در تکلم واضح 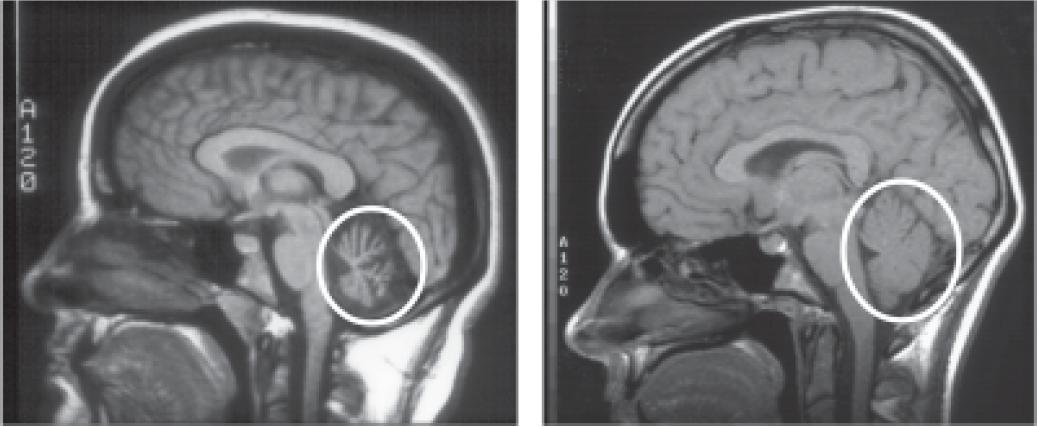

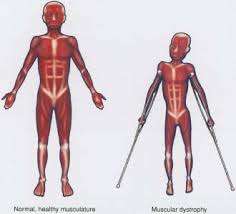 دوشن یک اختلال وابسته به کروموزوم X است که احتمال وقوع آن یک از هر 3500 نوزاد پسر است. تعداد مبتلایان به دیستروفی در ایران حدود 20 هزار نفر برآورد میشود که از این تعداد بین 15 تا 16 هزار نفر به دوشن مبتلا هستند. در حدود 100 پسر سالانه در انگلستان با عارضه DMD متولد میشوند. همیشه در حدود 1500 پسر با این نوع اختلال در انگلستان زندگی میکنند .بطور کلی در بین مردم احتمال تولد کودکان پسر با این نوع عارضه ،1 به 3500 است.این بیماری از نوع مغلوب وابسته به جنس می باشد. علت بیماریژن مسئول ساخت این دیستروفین( DMD)است و بر روی کروموزوم X قرار دارد .پروتئین دیستروفین اطراف سلول های ماهیچه ای برای محافظت ساختمان ماهیچه ساخته می شوند و مانع از خروج عناصر داخل سلول ماهیچه ای به فضای خارج از سلول می شود.بدون دیستروفین سلول ماهیچه ای قابل نفوذ خواهد بود و مواد بافت خارج سلولی وارد سلول ماهیچه شده و باعث تخریب و مرگ ماهیچه خواهد شد و در نهایت بافت چربی جای ماهیچه را می گیرد. علت عدم تولید پروتئین دیستروفین در ژن دی ام دی هنوز به طور قطع مشخص نیست ولی این عدم تولید ناشی از مسدود شدن مجاری تولید پروتئین دیسترفین می باشد. دیستروفی عضلانی دوشن یک بیماری ارثی مغلوب وابسته به X می باشد و همانند سایر بیماری های این گروه عموماً محدود به مردان شده و به ندرت در زنان دیده م یشود، مگر در مواردی نادر برای مثال حالتی که دختران حامل ژن DMD تنها واجد کروموزوم X (مبتلا به سندرم ترنر) باشند. یک سوم ژن های DMD در جمعیت، توسط مردان و دوسوم دیگر توسط زنان حامل، منتقل م یشود. اما از آنجا که سازش تولید مثلی مردان بیمار صفر است و قادر به انتقال بیماری به نسل بعد نیستند، بنابراین از میان بیماران دوشن یک سوم جهش یافته های جدید هستند و دوسوم سابقه فامیلی دارند .علایم ناشی از بیماری به دلیل طبیعت تدریجی و غافلگیرکننده بیماری ،تعیین دقیق سن شروع بیماری و ظهور علائم اولیه کار مشکلی است. والدین معمولاً از هیچ گونه ناهنجاری ،پیش از آنکه کودک آغاز به راه رفتن نماید، آگاه نیستند .یکی از علائمی که والدین را متوجه می سازد تأخیر کودک در راه رفتن است. دیگر نش انههای شروع بیماری راه رفتن اردکی شکل، افتادن های مکرر، راه رفتن روی پنجه پا ،ناتوانی در بالا رفتن از پله ها و ناتوانی در برخاستن از حالت نشسته می باشد.در مجموع و با توجه به آمارهای مختلف بهدست آمده، در 90 درصد بیماران شروع علائم بالینی بیش از 5سالگی است .مشخص ترین علامت در مراحل اولیه بیماری، بزرگ شدن عضلات اطراف شانه و زبان، عضلات ساعد و گاهی در عضلات دیگر نیز دیده م یشود. بزرگی عضلات در حقیقت به دلیل افزایش بافت همبند و چربی است که عضله را در حالت ضعف نگه م یدارد و به همین دلیل هم بزرگی کاذب نامیده می شود. این ویژگی در بیش از 95 درصد بیماران دیده می شود اما مختص این بیماری نیست و در برخی اشکال دیگر دیستروفی نیز یافت می شود. از دیگر علائم زود هنگام،علامت گاورس( Gowers sign) میباشد که نتیجه ضعف عضلات انبساطی چهارسر ران و زانو است .علامت گاورس نشانگر روش غیرمعمول این بیماران در برخاستن از حالت نشسته، به وسیله بالارفتن از پاهای آنها می باشد و معمولاً از 4تا 5 سالگی ظاهر م یگردد و از جمله نشانه های اولیه بیماری است با پیشرفت بیماری بالدار شدن کتف نیز آشکار م یگردد که این پدیده در نتیجه آتروفی ماهی چههای اطراف شانه است البته آشکارترین علامت زمانی است که پسران بیمار به ویلچر محدود می شوند. پسران مبتلا به دوشن در 20 درصد موارد IQ کمتر از 70 دارند. بزرگی پشت ساق پا( enlarged calve) در اکثر مبتلایان به دوشن و همچنین بکر مشاهده میگردد .روش تشخیص تشخیص اولیه بیماری به وسیله علائم بالینی و الگوی وراثتی، انجام می گیرد. اما باید توجه داشت بهدلیل ویژگی های مشابهی که میان این بیماری و بعضی بیمار یهای دیگر وجود دارد، این تشخیص قطعی نخواهد بود و تأیید این تشخیص به وسیله تعیین سطح آنزیم کراتین کیناز سرمی ،الکترومیوگرافی، نمونه برداری از عضله و نیز آزمایشات مولکولی انجام م یگیرد .یکبار که نوزادی با عارضه DMD در خانواده ای متولد شده باشد، اغلب در تشخیصهای پیش از زایمان برای بارداریهای بعدی هم برای مادر و هم برای دیگر زنانی که احتمال ناقل بودن در آنها داده شود مشاوره صورت می گیرد. بطور طبیعی این امکان وجود دارد که توسط اطلاعات دقیق بدست آمده از مطالعه DNA، وضعیت جنین تشخیص داده شود. با بررسی انجام شده بر روی DNA جنین توسط نمونه برداری از پرده بیرونی آن میتوان به این اطلاعات پی برد. این آزمایش بر روی تکه ای از پوششی که در هفته 11 الی 21 بارداری اطراف جنین قرار دارد انجام میشود.درمان تاکنون هیچ درمانی برای دیستروفی ماهیچه ای دوشن یافت نشده است اگرچه اخیراً تحقیقات نشان میدهد به وسیله سلول های بنیادی می توان بافت ماهیچه های سالم را جایگزین بافت ماهیچه ای آسیب دیده کرد. عفونت های ریوی را باید به سرعت درمان نمود. در این بیماران، فیزیوتراپی می تواند در به تعویق افتادن سستی عضلات مچ پاها، مفصل ران و آرنج مؤثر باشد. در حال حاضر درمان داروئی و ژن درمانی تحت بررسی است. تنها با فیزیوتراپی و کاردرمانی می توان از انحراف ستون فقرات و قفسه سینه که موجب جمع شدگی بدن میشود جلوگیری نمود از میان داروهای متعددی که مورد مطالعه و بررسی قرار گرفته، تنها داروئی که هنوز مصرف م یشود، پرودنیزون است .مراقبت های لازم نارسایی قلبی و ذات الریه مهمترین عامل مرگ این بیماران است که بدون ک مکهای جانبی بین 13 تا 19 سالگی رخ میدهد .کمی بعد از روزهای اولیه تشخیص، نرمشهای مداوم مهم هستند اما لزوما به نظارت پزشکی نیاز نیست، اگر چه ممکن است احساس شود ارتباط با یک فیزوتراپیست می تواند مفید باشد. در این مرحله مهمترین راهی که یک پزشک میتواند به شما به عنوان والدین کمک کند، تا حد امکان آموزش در مورد دیستروفی عضلات و تدارک و تهیه مشاوره های ژنتیکی است. همچنین این امکان وجود دارد که در این مرحله برای مدت طولانی و برای کمک و مشاوره مستمر مجموعه برنامه ای رادرنظرگرفت. پیش آگهیتا سنین تقریبا 8 الی 11 سالگی(بندرت سنین قبل و یا بعد) پسران برای راه رفتن ناتوان میشوند و امید به زندگی در آنها بسختی تا اواخر نوجوانی یا بیست سالگی میرسد .همچنین در نتیجه ضعف عضلات بین دنده ای، مشکلات تنفسی شدت می یابد .درگیری عضلات تنفسی معمولاً بهصورت سرفه ضعیف، عفونت های مکرر تنفسی، و کاهش ذخیره تنفسی ظاهر م یشود. نارسائی تنفسی در خواب، ذات الریه و گاهی آسپیراسیون و انسداد راه های هوائی علت مرگ بیماران است. شایعترین علت مرگ این بیماران عفونت های ریوی م یباشد. پسران حامل ژن بیماری دوشن از اوایل نوزادی از عوارض این بیماری رنج می برند. هنگامی که این پسران به سن بلوغ می رسند بافت عضلانی کاملا از بین میرود و فلج و سپس مرگ اتفاق میافتد.
دوشن یک اختلال وابسته به کروموزوم X است که احتمال وقوع آن یک از هر 3500 نوزاد پسر است. تعداد مبتلایان به دیستروفی در ایران حدود 20 هزار نفر برآورد میشود که از این تعداد بین 15 تا 16 هزار نفر به دوشن مبتلا هستند. در حدود 100 پسر سالانه در انگلستان با عارضه DMD متولد میشوند. همیشه در حدود 1500 پسر با این نوع اختلال در انگلستان زندگی میکنند .بطور کلی در بین مردم احتمال تولد کودکان پسر با این نوع عارضه ،1 به 3500 است.این بیماری از نوع مغلوب وابسته به جنس می باشد. علت بیماریژن مسئول ساخت این دیستروفین( DMD)است و بر روی کروموزوم X قرار دارد .پروتئین دیستروفین اطراف سلول های ماهیچه ای برای محافظت ساختمان ماهیچه ساخته می شوند و مانع از خروج عناصر داخل سلول ماهیچه ای به فضای خارج از سلول می شود.بدون دیستروفین سلول ماهیچه ای قابل نفوذ خواهد بود و مواد بافت خارج سلولی وارد سلول ماهیچه شده و باعث تخریب و مرگ ماهیچه خواهد شد و در نهایت بافت چربی جای ماهیچه را می گیرد. علت عدم تولید پروتئین دیستروفین در ژن دی ام دی هنوز به طور قطع مشخص نیست ولی این عدم تولید ناشی از مسدود شدن مجاری تولید پروتئین دیسترفین می باشد. دیستروفی عضلانی دوشن یک بیماری ارثی مغلوب وابسته به X می باشد و همانند سایر بیماری های این گروه عموماً محدود به مردان شده و به ندرت در زنان دیده م یشود، مگر در مواردی نادر برای مثال حالتی که دختران حامل ژن DMD تنها واجد کروموزوم X (مبتلا به سندرم ترنر) باشند. یک سوم ژن های DMD در جمعیت، توسط مردان و دوسوم دیگر توسط زنان حامل، منتقل م یشود. اما از آنجا که سازش تولید مثلی مردان بیمار صفر است و قادر به انتقال بیماری به نسل بعد نیستند، بنابراین از میان بیماران دوشن یک سوم جهش یافته های جدید هستند و دوسوم سابقه فامیلی دارند .علایم ناشی از بیماری به دلیل طبیعت تدریجی و غافلگیرکننده بیماری ،تعیین دقیق سن شروع بیماری و ظهور علائم اولیه کار مشکلی است. والدین معمولاً از هیچ گونه ناهنجاری ،پیش از آنکه کودک آغاز به راه رفتن نماید، آگاه نیستند .یکی از علائمی که والدین را متوجه می سازد تأخیر کودک در راه رفتن است. دیگر نش انههای شروع بیماری راه رفتن اردکی شکل، افتادن های مکرر، راه رفتن روی پنجه پا ،ناتوانی در بالا رفتن از پله ها و ناتوانی در برخاستن از حالت نشسته می باشد.در مجموع و با توجه به آمارهای مختلف بهدست آمده، در 90 درصد بیماران شروع علائم بالینی بیش از 5سالگی است .مشخص ترین علامت در مراحل اولیه بیماری، بزرگ شدن عضلات اطراف شانه و زبان، عضلات ساعد و گاهی در عضلات دیگر نیز دیده م یشود. بزرگی عضلات در حقیقت به دلیل افزایش بافت همبند و چربی است که عضله را در حالت ضعف نگه م یدارد و به همین دلیل هم بزرگی کاذب نامیده می شود. این ویژگی در بیش از 95 درصد بیماران دیده می شود اما مختص این بیماری نیست و در برخی اشکال دیگر دیستروفی نیز یافت می شود. از دیگر علائم زود هنگام،علامت گاورس( Gowers sign) میباشد که نتیجه ضعف عضلات انبساطی چهارسر ران و زانو است .علامت گاورس نشانگر روش غیرمعمول این بیماران در برخاستن از حالت نشسته، به وسیله بالارفتن از پاهای آنها می باشد و معمولاً از 4تا 5 سالگی ظاهر م یگردد و از جمله نشانه های اولیه بیماری است با پیشرفت بیماری بالدار شدن کتف نیز آشکار م یگردد که این پدیده در نتیجه آتروفی ماهی چههای اطراف شانه است البته آشکارترین علامت زمانی است که پسران بیمار به ویلچر محدود می شوند. پسران مبتلا به دوشن در 20 درصد موارد IQ کمتر از 70 دارند. بزرگی پشت ساق پا( enlarged calve) در اکثر مبتلایان به دوشن و همچنین بکر مشاهده میگردد .روش تشخیص تشخیص اولیه بیماری به وسیله علائم بالینی و الگوی وراثتی، انجام می گیرد. اما باید توجه داشت بهدلیل ویژگی های مشابهی که میان این بیماری و بعضی بیمار یهای دیگر وجود دارد، این تشخیص قطعی نخواهد بود و تأیید این تشخیص به وسیله تعیین سطح آنزیم کراتین کیناز سرمی ،الکترومیوگرافی، نمونه برداری از عضله و نیز آزمایشات مولکولی انجام م یگیرد .یکبار که نوزادی با عارضه DMD در خانواده ای متولد شده باشد، اغلب در تشخیصهای پیش از زایمان برای بارداریهای بعدی هم برای مادر و هم برای دیگر زنانی که احتمال ناقل بودن در آنها داده شود مشاوره صورت می گیرد. بطور طبیعی این امکان وجود دارد که توسط اطلاعات دقیق بدست آمده از مطالعه DNA، وضعیت جنین تشخیص داده شود. با بررسی انجام شده بر روی DNA جنین توسط نمونه برداری از پرده بیرونی آن میتوان به این اطلاعات پی برد. این آزمایش بر روی تکه ای از پوششی که در هفته 11 الی 21 بارداری اطراف جنین قرار دارد انجام میشود.درمان تاکنون هیچ درمانی برای دیستروفی ماهیچه ای دوشن یافت نشده است اگرچه اخیراً تحقیقات نشان میدهد به وسیله سلول های بنیادی می توان بافت ماهیچه های سالم را جایگزین بافت ماهیچه ای آسیب دیده کرد. عفونت های ریوی را باید به سرعت درمان نمود. در این بیماران، فیزیوتراپی می تواند در به تعویق افتادن سستی عضلات مچ پاها، مفصل ران و آرنج مؤثر باشد. در حال حاضر درمان داروئی و ژن درمانی تحت بررسی است. تنها با فیزیوتراپی و کاردرمانی می توان از انحراف ستون فقرات و قفسه سینه که موجب جمع شدگی بدن میشود جلوگیری نمود از میان داروهای متعددی که مورد مطالعه و بررسی قرار گرفته، تنها داروئی که هنوز مصرف م یشود، پرودنیزون است .مراقبت های لازم نارسایی قلبی و ذات الریه مهمترین عامل مرگ این بیماران است که بدون ک مکهای جانبی بین 13 تا 19 سالگی رخ میدهد .کمی بعد از روزهای اولیه تشخیص، نرمشهای مداوم مهم هستند اما لزوما به نظارت پزشکی نیاز نیست، اگر چه ممکن است احساس شود ارتباط با یک فیزوتراپیست می تواند مفید باشد. در این مرحله مهمترین راهی که یک پزشک میتواند به شما به عنوان والدین کمک کند، تا حد امکان آموزش در مورد دیستروفی عضلات و تدارک و تهیه مشاوره های ژنتیکی است. همچنین این امکان وجود دارد که در این مرحله برای مدت طولانی و برای کمک و مشاوره مستمر مجموعه برنامه ای رادرنظرگرفت. پیش آگهیتا سنین تقریبا 8 الی 11 سالگی(بندرت سنین قبل و یا بعد) پسران برای راه رفتن ناتوان میشوند و امید به زندگی در آنها بسختی تا اواخر نوجوانی یا بیست سالگی میرسد .همچنین در نتیجه ضعف عضلات بین دنده ای، مشکلات تنفسی شدت می یابد .درگیری عضلات تنفسی معمولاً بهصورت سرفه ضعیف، عفونت های مکرر تنفسی، و کاهش ذخیره تنفسی ظاهر م یشود. نارسائی تنفسی در خواب، ذات الریه و گاهی آسپیراسیون و انسداد راه های هوائی علت مرگ بیماران است. شایعترین علت مرگ این بیماران عفونت های ریوی م یباشد. پسران حامل ژن بیماری دوشن از اوایل نوزادی از عوارض این بیماری رنج می برند. هنگامی که این پسران به سن بلوغ می رسند بافت عضلانی کاملا از بین میرود و فلج و سپس مرگ اتفاق میافتد. 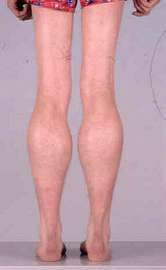

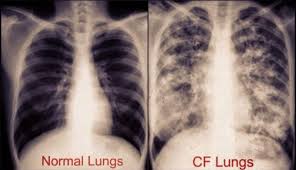 شایع ترین اختلال اتوزومی مغلوب در کودکان اروپایی 1 در هر 2500 تولد است. فیبروز کیستی با میزان بروز 1 مورد در هر 3200 تولد زنده در ایالات متحده، فراوانترین بیماری کشنده ژنتیکی است که جمعیت قفقازی را مبتلا میکند. در حدود یک نفر از هر 25 نفر در ایالات متحده ی آمریکا حامل هترو زیگوت برای الل جهش یافته ی سیستیک فیبروزیس است. این بیماری در بین آسیایی ها 1مورد در 32000 تولد زنده و آمریکایی های آفریقایی تبار 1 مورد در 15000 تولد زنده ناشایع است.فراوانی CF در بین متولدین سفید پوست برابر با 1به25000است. فراوانی حاملین CFبرابر با 1 به 25 است. اگر هم مادر و هم پدر ازخانوادهای باشندکه فیبروز کیستیک در آن وجود داشته است ، شانس بروز بی ماری در کودک آنها 1به 4است.علت بیماری نوزاد از بدو تولد این بیماری را از پدر و مادر خود به ارث می برد. اگر پدر و مادر هر دو سالم باشند اما هر یک از آنها حامل یک ژن معیوب فیبروز کیستی باشند هر فرزندی را که مادر حامله شود 25درصد شانس به ارث بردن هر دو ژن معیوب و در نتیجه ابتلاء به فیبروز کیستی را دارد علت اصلی این بیماری، نقص در ژن کدکننده پروتئین RTFC است .علایم ناشی از بیماری ترشحات غیر طبیعی از علایم اصلی این اختلال است که شدیدترین اثر آن در سیستم تنفسی است که موکوس چسبنده ای به صورت غیر طبیعی راه های طبیعی هوایی را مسدود می کند. مژک هایی که نایژه ها را مفروش می کنند به آسانی نمی توانند موکوس را بر دارند بنابراین به محیطی برای رشد باکتری های خطرناک تبدیل می شوند. این باکتری ها یا سموم آن ها به بافتهای اطراف آسیب رسانده و منجر به ذات الریه عود کننده و مشکلات دیگری می شوند در این حالت، غده های برونشی بجای تولید مخاط رقیق طبیعی، خلط غلیظ و لزجی تولید می کنند که منجر به بسته شدن مجاری عبور هوا میشود و در نهایت عفونتهای ریوی را بوجود میآورد و وقتیکه بخشهایکوچکی از ریهها فرسوده شوند خطر ذات الریه را به همراه دارد. این عارضه اخیر و عفونتهای عود کننده در مبتلایان به فیبروزکیستی شایع است .عفونتهای عود کننده همراه با سرفه و ناراحتیهای تنفسی اسهال، ممکن است بصورت دوره ای با یبوست جایش را عوض کند. مدفوع چرب و بدبو نارسایی رشد ورم شکم و تحلیل رفتن دستها و پاها .در رودههای کودک مبتلا به فیبروز کیستی ،لوزالمعده نمی تواند برخی آنزیمهای حیاتی را تولید کند .این آنزیمها به عمل هضم کمک و غذا را شکسته و تجزیه میکنند تا بدن بتواند آنها را آسانتر جذب کند. فقدان این آنزیم های هضم کننده بدان معنا است که غذا بدرستی جذب نمیشود و موجب اسهال و مدفوع بدبو می گردد. از آنجائیکه غذا جذب نمیشود بدن بسیاری از مواد غذایی ضروری را که برای سلامتی لازم است دریافت نمیکند در نتیجه ،کودک رشد نمیکند و همچنان کوچک و کم وزن باقی میماند ممکن است بعد از هر دوره اسهال، یبوست جایگزین آن شود و توالی این مطلب میتواند عملاً روده را مسدود کند.روش تشخیص اگر چه آزمایشهای ساده ای روی خون و مدفوع نوزادان می توان انجام داد اما آزمایش قطعی برای فیبروز کیستی تست عرق می باشد زیرا میزان نمک عرق در این بیماران بالا است در مورد برادران و خواهران فرد مبتلا و در نوزادانی که دچار مراحل عود کننده ذات الریه هستند و دچار توقف رشد شده اند نیز این آزمایش انجام میگیرد. تهیه نمونه ها از مایع آمنیون و خون با استفاده از روش های روتین استخراج ژنوم و بررسی برای یافتن یکی از رایج ترین جهش ها در ژن CF با استفاده از روش 1RFLP انجام میگیرد.درمان درمان های کنونی برای سیستیک فیبروزیس( CF)عموما جهت تسکین علایم بیماران هستند. آنتی بیوتی کها برای کنترل عفونتهای باکتریایی مورد استفاده قرار می گیرند اما درمان فیزیکی روزانه برای پاک کردن موک وسها از سیستم تنفسی مورد نیاز است.به دلیل بروز عفونت های باکتریایی مزمن و کشنده در راههای تنفسی کنترل عفونتو حفظ عملکرد تنفسی مطلوب کلید اصلی درمان است.دارو درمانی در این بیماران شامل موارد زیر است:آنتی بیوتیک ها به منظور پیشگیری از عفونت استفاده می شوند .داروهای گشاد کننده برونش برای باز نگه داشتن مسیرهای تنفسی استفاده میشود.از کورتون ها به منظور کاهش التهاب در سیستم تنفسی استفاده میگردد.برحسب نیاز از آنزیمهای پانکراس(لوزالمعده) برای کاهش مشکلات گوارشی استفاده میشود.مکمل های ویتامین های محلول در چربی برای بیمار تجویز میشود که معمولا به صورت روزانه مصرف میشود.زمانی که کودک در بیمارستان بستری است یا در موارد بیماری ریوی پیشرفته برای پیشگیری از کمبود اکسیژن ممکن است اکسیژن درمانی انجام شود .مراقبت های لازم رویکرد تیمی برای کم ک به مراقب ت از کودک(درمانگر تنفسی، پرستار، متخصص تغذیه، فیریوتراپ ،مشاور، مددکار اجتماعی)تا جایی که میتوانید اطلاعات خود را در مورد این بیماری بالا ببرید. توجه داشته باشید که رژیم غذایی ، دارو، و تشخیص زودهنگام عفونت بسیار مهم هستند.تخلیه روزانه مخاط ریه ها با تغییر وضعی ت بدن و نیز ضربه زدن به قفسه سی نه برای تکان دادن و نرم کردن تکه های مخاطی چسبنا ک، در بهبود علایم ظاهری بیمار مؤثر خواهد بود .هر گاه کود ک علایم تنفسی دارد ،از دستگاه بخور استفاده کنید. رطوبت به ناز ک کردن مخاط کمک می کند و بنابراین مخاط راحت تر با سرفه خارج می شود. دستگاه بخور را هر روز تمیز کنید .واکسیناسیون کودک خود را به روز نگاه دارید. کودک باید واکسن آنفلوانزا نیز دریافت کند .کودک خود را تشویق کنید تا زندگی طبیعی و فعال داشته باشد .پیوند ریه نیز تدریجاً به جرگه روشهای درمانی پیوسته است.پیش آگهیتغییرات آناتومیک در CF بسیار متغییر هستند و به این بستگی دارد که کدام غده و با چه شدتی درگیر شده است.اختلالات پانکراسی در تقریبا 85 تا 90 درصد بیماران مبتلا به CF وجود دارد. در موارد خفیف تر فقط تجمع موکوس در مجاری کوچک اتساع یافته غدد اگزو کرین وجود دارد. در موارد پیشرفته تر که معمولا در بچه های بزرگتر یا نوجوانان دیده میشود، مجاری کاملا مسدود شده اند که این حالت باعث آتروفی غدد برون ریز و فیبروز پیشرونده میگردد. آزواسپرمی و ناباروری در 95 درصد مردانی که تا بزرگسالی زنده می مانند دیده می شود.فقدان دو طرفه وازودفران یافته ی شایعی در این بیماران می باشد این بی ماری در حال حاضر علاج ناپذیر است و غالباً بیمار در همان بچگی فوت می کند. مراقبت دقیق و درازمدت توسط والدین و تیم پزشکی به کودک کمک می کند تا زندگی تقریباً راحتی داشته باشد. البته کودکانی که دچار انواع خفیف تر بیماری هستند تا دوران بزرگسالی زنده می مانند ،خصوصاً اگر این بیماری زود تشخیص داده شود .میانگین طول عمر بیماران 28 سال است . محققان ژن مسؤول بیماری فیبروز کیستیک را کشف کرده اند. هم اکنون در مورد روش های نوین پیشگیری و درمان این بیماری کارهای زیادی در حال انجام است.
شایع ترین اختلال اتوزومی مغلوب در کودکان اروپایی 1 در هر 2500 تولد است. فیبروز کیستی با میزان بروز 1 مورد در هر 3200 تولد زنده در ایالات متحده، فراوانترین بیماری کشنده ژنتیکی است که جمعیت قفقازی را مبتلا میکند. در حدود یک نفر از هر 25 نفر در ایالات متحده ی آمریکا حامل هترو زیگوت برای الل جهش یافته ی سیستیک فیبروزیس است. این بیماری در بین آسیایی ها 1مورد در 32000 تولد زنده و آمریکایی های آفریقایی تبار 1 مورد در 15000 تولد زنده ناشایع است.فراوانی CF در بین متولدین سفید پوست برابر با 1به25000است. فراوانی حاملین CFبرابر با 1 به 25 است. اگر هم مادر و هم پدر ازخانوادهای باشندکه فیبروز کیستیک در آن وجود داشته است ، شانس بروز بی ماری در کودک آنها 1به 4است.علت بیماری نوزاد از بدو تولد این بیماری را از پدر و مادر خود به ارث می برد. اگر پدر و مادر هر دو سالم باشند اما هر یک از آنها حامل یک ژن معیوب فیبروز کیستی باشند هر فرزندی را که مادر حامله شود 25درصد شانس به ارث بردن هر دو ژن معیوب و در نتیجه ابتلاء به فیبروز کیستی را دارد علت اصلی این بیماری، نقص در ژن کدکننده پروتئین RTFC است .علایم ناشی از بیماری ترشحات غیر طبیعی از علایم اصلی این اختلال است که شدیدترین اثر آن در سیستم تنفسی است که موکوس چسبنده ای به صورت غیر طبیعی راه های طبیعی هوایی را مسدود می کند. مژک هایی که نایژه ها را مفروش می کنند به آسانی نمی توانند موکوس را بر دارند بنابراین به محیطی برای رشد باکتری های خطرناک تبدیل می شوند. این باکتری ها یا سموم آن ها به بافتهای اطراف آسیب رسانده و منجر به ذات الریه عود کننده و مشکلات دیگری می شوند در این حالت، غده های برونشی بجای تولید مخاط رقیق طبیعی، خلط غلیظ و لزجی تولید می کنند که منجر به بسته شدن مجاری عبور هوا میشود و در نهایت عفونتهای ریوی را بوجود میآورد و وقتیکه بخشهایکوچکی از ریهها فرسوده شوند خطر ذات الریه را به همراه دارد. این عارضه اخیر و عفونتهای عود کننده در مبتلایان به فیبروزکیستی شایع است .عفونتهای عود کننده همراه با سرفه و ناراحتیهای تنفسی اسهال، ممکن است بصورت دوره ای با یبوست جایش را عوض کند. مدفوع چرب و بدبو نارسایی رشد ورم شکم و تحلیل رفتن دستها و پاها .در رودههای کودک مبتلا به فیبروز کیستی ،لوزالمعده نمی تواند برخی آنزیمهای حیاتی را تولید کند .این آنزیمها به عمل هضم کمک و غذا را شکسته و تجزیه میکنند تا بدن بتواند آنها را آسانتر جذب کند. فقدان این آنزیم های هضم کننده بدان معنا است که غذا بدرستی جذب نمیشود و موجب اسهال و مدفوع بدبو می گردد. از آنجائیکه غذا جذب نمیشود بدن بسیاری از مواد غذایی ضروری را که برای سلامتی لازم است دریافت نمیکند در نتیجه ،کودک رشد نمیکند و همچنان کوچک و کم وزن باقی میماند ممکن است بعد از هر دوره اسهال، یبوست جایگزین آن شود و توالی این مطلب میتواند عملاً روده را مسدود کند.روش تشخیص اگر چه آزمایشهای ساده ای روی خون و مدفوع نوزادان می توان انجام داد اما آزمایش قطعی برای فیبروز کیستی تست عرق می باشد زیرا میزان نمک عرق در این بیماران بالا است در مورد برادران و خواهران فرد مبتلا و در نوزادانی که دچار مراحل عود کننده ذات الریه هستند و دچار توقف رشد شده اند نیز این آزمایش انجام میگیرد. تهیه نمونه ها از مایع آمنیون و خون با استفاده از روش های روتین استخراج ژنوم و بررسی برای یافتن یکی از رایج ترین جهش ها در ژن CF با استفاده از روش 1RFLP انجام میگیرد.درمان درمان های کنونی برای سیستیک فیبروزیس( CF)عموما جهت تسکین علایم بیماران هستند. آنتی بیوتی کها برای کنترل عفونتهای باکتریایی مورد استفاده قرار می گیرند اما درمان فیزیکی روزانه برای پاک کردن موک وسها از سیستم تنفسی مورد نیاز است.به دلیل بروز عفونت های باکتریایی مزمن و کشنده در راههای تنفسی کنترل عفونتو حفظ عملکرد تنفسی مطلوب کلید اصلی درمان است.دارو درمانی در این بیماران شامل موارد زیر است:آنتی بیوتیک ها به منظور پیشگیری از عفونت استفاده می شوند .داروهای گشاد کننده برونش برای باز نگه داشتن مسیرهای تنفسی استفاده میشود.از کورتون ها به منظور کاهش التهاب در سیستم تنفسی استفاده میگردد.برحسب نیاز از آنزیمهای پانکراس(لوزالمعده) برای کاهش مشکلات گوارشی استفاده میشود.مکمل های ویتامین های محلول در چربی برای بیمار تجویز میشود که معمولا به صورت روزانه مصرف میشود.زمانی که کودک در بیمارستان بستری است یا در موارد بیماری ریوی پیشرفته برای پیشگیری از کمبود اکسیژن ممکن است اکسیژن درمانی انجام شود .مراقبت های لازم رویکرد تیمی برای کم ک به مراقب ت از کودک(درمانگر تنفسی، پرستار، متخصص تغذیه، فیریوتراپ ،مشاور، مددکار اجتماعی)تا جایی که میتوانید اطلاعات خود را در مورد این بیماری بالا ببرید. توجه داشته باشید که رژیم غذایی ، دارو، و تشخیص زودهنگام عفونت بسیار مهم هستند.تخلیه روزانه مخاط ریه ها با تغییر وضعی ت بدن و نیز ضربه زدن به قفسه سی نه برای تکان دادن و نرم کردن تکه های مخاطی چسبنا ک، در بهبود علایم ظاهری بیمار مؤثر خواهد بود .هر گاه کود ک علایم تنفسی دارد ،از دستگاه بخور استفاده کنید. رطوبت به ناز ک کردن مخاط کمک می کند و بنابراین مخاط راحت تر با سرفه خارج می شود. دستگاه بخور را هر روز تمیز کنید .واکسیناسیون کودک خود را به روز نگاه دارید. کودک باید واکسن آنفلوانزا نیز دریافت کند .کودک خود را تشویق کنید تا زندگی طبیعی و فعال داشته باشد .پیوند ریه نیز تدریجاً به جرگه روشهای درمانی پیوسته است.پیش آگهیتغییرات آناتومیک در CF بسیار متغییر هستند و به این بستگی دارد که کدام غده و با چه شدتی درگیر شده است.اختلالات پانکراسی در تقریبا 85 تا 90 درصد بیماران مبتلا به CF وجود دارد. در موارد خفیف تر فقط تجمع موکوس در مجاری کوچک اتساع یافته غدد اگزو کرین وجود دارد. در موارد پیشرفته تر که معمولا در بچه های بزرگتر یا نوجوانان دیده میشود، مجاری کاملا مسدود شده اند که این حالت باعث آتروفی غدد برون ریز و فیبروز پیشرونده میگردد. آزواسپرمی و ناباروری در 95 درصد مردانی که تا بزرگسالی زنده می مانند دیده می شود.فقدان دو طرفه وازودفران یافته ی شایعی در این بیماران می باشد این بی ماری در حال حاضر علاج ناپذیر است و غالباً بیمار در همان بچگی فوت می کند. مراقبت دقیق و درازمدت توسط والدین و تیم پزشکی به کودک کمک می کند تا زندگی تقریباً راحتی داشته باشد. البته کودکانی که دچار انواع خفیف تر بیماری هستند تا دوران بزرگسالی زنده می مانند ،خصوصاً اگر این بیماری زود تشخیص داده شود .میانگین طول عمر بیماران 28 سال است . محققان ژن مسؤول بیماری فیبروز کیستیک را کشف کرده اند. هم اکنون در مورد روش های نوین پیشگیری و درمان این بیماری کارهای زیادی در حال انجام است. 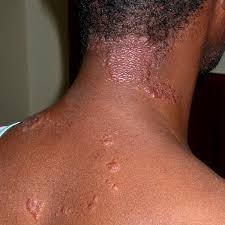
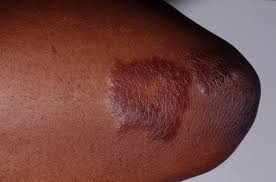 این بیماری یک بیماری التهابی سیستمیک است که به علت نامشخص ایجاد میگردد در سراسر دنیا از هر 100000 نفر20 نفر به سارکوئیدوز مبتلا میشود و این در حالی است که ژاپنی ها بخصوص، زنان ژاپنی درسنین بالای40 سال نسبت به سایر جمعیت دنیا ریسک ابتلا به سارکوئیدوز و بخصوص سارکوئیدوز قلبی دارند. عمومی ترین تظاهرات آن در 80% موارد ریوی است اما می تواند با درگیری سایر ارگانها مانند پوست، غدد پاروتید، طحال ،کبد، درگیری سیستم اعصاب مرکزی، استخوان و یووئیت، غدد لنفاوی و قلب نیز تظاهر یابد. در 50% بیماران مبتلا به سارکوئیدوز درگیری قلب نیز دیده شده است. زنان قدری بیش از مردان مبتلا می شوند .تخمین زده می شود شیوع آن در محدوده 1 تا40 مورد در هر 100000 نفر باشد میزان شیوع سالانه تنظیم شده براساس سن 5/35 مورد در هر 100000 نفر برای آفریقایی آمریکاییها و 9/10در هر 100000 مورد برای سفیدپوستان است.علایم ناشی از بیماری درگیری موضعی قلب یکی از شدیدترین تظاهرات سارکوئیدوز بوده و ممکن است سبب تهدید زندگی فرد ومرگ وی گردد .علائم بالینی سارکوئیدوز قلبی بستگی به محل و وسعت ضایعات التهابی گرنولوماتو دارد و می تواند با تظاهرات آریتمی، اختلالات هدایتی و نارسایی احتقانی قلب خود را نشان دهد.سارکوئیدوز یک بیماری سیستمیک است که با ایجاد گرانولومهای بدون کازئوم به علت یا به عللی که هنوز دقیقا شناخته نشده اند ،مشخص می شود. به این جهت، برخلاف بیماری هایی که منشاء آنها معلوم است، در تعریف آن با اشکال روبه رو می شویم .انجمن بیماری های ریه آمریکا همراه با انجمن بیماری های ریه اروپا و انجمن جهانی سارکوئیدوز تعریف توصیفی زیر را پیشنهاد کرده اند. سارکوئیدوز یک اختلال چند ارگانی سیستم به علت (یا علل) ناشناخته است. معمولا جوانان یا بالغین میان سال را مبتلا می کند و غالبا با لنفادنوپاتی در ناف ریتین و انفیلتراسیون در ریه و ضایعات پوست و چشم تظاهر می کند. کبد، طحال، غدد لنفی ،غدد بزاق، قلب، سیستم عصبی، عضلات، استخوان ها و سایر اعضاء نیز ممکن است مبتلا شوند.آغاز سارکوئیدوز ممکن است به صورت حاد و آشکار و یا نهفته و با تظاهرات خفیف همراه باشد. شکل حاد با علایم عمومی و موضعی شدید بروز میکند و سیر آن معمولا کوتاهتر(چند ماه یا حدود یک سال)است و خود به خود با درمان به طرف بهبود و ضایعات و یا به طرف ازمان تثبیت میرود.یافته های بالینی دیگر شامل کاهش حجم اجبار بازدمی ،ظرفیت حیاتی، و ظرفیت انتظار هستند. آنمی، لنفوسیتوپنی ،بالارفتن میزان رسوب اریتروسیت ،افزایش تست های عملکرد کبدی ،هایپرکلسمی، و نفروپاتی هایپرکلسمیک نیز از یافته های معمول هستند. غدد لنفاوی آنها ممکن است متورم شده و شما متوجه توده هایی در زیر بازو، در گردن و یا در کشاله ران شوید. پزشک ممکن است متوجه تورم غدد لنفاوی در ناحیه قفسه سینه در زمان مشاهده عکس قفسه سینه شما بشود .پوست: شما ممکن است توده های کوچکی را درست درزیر پوستتان حبس کنید. همچنین ممکن است یک راش (لکه)ارغوانی رنگ و برجسته بر روی بینی، گونه ها، چانه و گوشها ایجاد شود. به این لکه ،lupus pernio گفته می شود.اریتم نودوزا نیز حالتی است که باعث ایجاد توده های قرمز و گردی اغلب در ساق پا می شود. این مساله ممکن است در واقع شروع بیماری سارکوئیدوز باشد و شایعترین نوع ضایعات پوستی در سارکوئیدوز می باشد.چشمها: سارکوئیدوز می تواند باعث التهاب چشمها و در نتیجه تاثیر بر روی دید شما شود. چشمها معمولا قرمز و دردناک میشوند. اگر شما مبتلا به سارکوئیدوز هستید و علایم چشمی در شما بروز کرد، حتما به پزشک مراجعه کنیدقلب: سارکوئیدوز باعث کند شدن کار قلب و یا منظم شدن ان می شود. آسیب به ریه ها که به علت سارکوئیدوز ایجاد شده می تواند تغییراتی در سمت راست قلب ایجاد کرده و آن را بزرگ نماید. این مساله می تواند در صورت عدم درمان باعث نارسایی سمت چپ قلب نیز شود. معمولا سارکوئیدوز باعث بزرگی قلب میشود که به آن کاردیومیوپاتی می گویند. این مساله به این معناست که قلب شما نمی تواند مثل قبل، قوی و موثر کار کند و به همین علت شما دچار تنگی نفس می شوید .اعصاب: سیستم عصبی شما ممکن است به چندین روش در سارکوئیدوز درگیر میشود. این بیماری می تواند باعث بروز سردرد ،مشکل در بلع، افتادگی صورت، نوعی مننژیت و مشکلات بینایی و شنوایی گردد. همچنین ممکن است بی حسی، احساس مورمور کردن و خواب رفتگی در صورت، دستها و پاهای شما ایجاد شود. در موارد نادر حتی ممکن است تشنج و یا سکته نیز بروز کند.کلیه ها : سارکوئیدوز تاثیر زیادی بر روی کلیه ها دارد .انسداد مجاری اشک و ضایعات آن باعث اشک ریزش می شود. بیمار ممکن است از آنوسمی شکایت داشته باشد.عوارض ناشی از این بیماری شاملعفونت قارچی ریهآب سیاه و کوری چشم )به ندرت(سنگ کلیه به علت افزایش کلسیم خون و یا ادرارپوکی استخوان و دیگر عوارض ناشی از مصرف کورتیکواستروئیدها برای مدت زمان طولانیپرفشاری خون ریویناباروری در مردان
این بیماری یک بیماری التهابی سیستمیک است که به علت نامشخص ایجاد میگردد در سراسر دنیا از هر 100000 نفر20 نفر به سارکوئیدوز مبتلا میشود و این در حالی است که ژاپنی ها بخصوص، زنان ژاپنی درسنین بالای40 سال نسبت به سایر جمعیت دنیا ریسک ابتلا به سارکوئیدوز و بخصوص سارکوئیدوز قلبی دارند. عمومی ترین تظاهرات آن در 80% موارد ریوی است اما می تواند با درگیری سایر ارگانها مانند پوست، غدد پاروتید، طحال ،کبد، درگیری سیستم اعصاب مرکزی، استخوان و یووئیت، غدد لنفاوی و قلب نیز تظاهر یابد. در 50% بیماران مبتلا به سارکوئیدوز درگیری قلب نیز دیده شده است. زنان قدری بیش از مردان مبتلا می شوند .تخمین زده می شود شیوع آن در محدوده 1 تا40 مورد در هر 100000 نفر باشد میزان شیوع سالانه تنظیم شده براساس سن 5/35 مورد در هر 100000 نفر برای آفریقایی آمریکاییها و 9/10در هر 100000 مورد برای سفیدپوستان است.علایم ناشی از بیماری درگیری موضعی قلب یکی از شدیدترین تظاهرات سارکوئیدوز بوده و ممکن است سبب تهدید زندگی فرد ومرگ وی گردد .علائم بالینی سارکوئیدوز قلبی بستگی به محل و وسعت ضایعات التهابی گرنولوماتو دارد و می تواند با تظاهرات آریتمی، اختلالات هدایتی و نارسایی احتقانی قلب خود را نشان دهد.سارکوئیدوز یک بیماری سیستمیک است که با ایجاد گرانولومهای بدون کازئوم به علت یا به عللی که هنوز دقیقا شناخته نشده اند ،مشخص می شود. به این جهت، برخلاف بیماری هایی که منشاء آنها معلوم است، در تعریف آن با اشکال روبه رو می شویم .انجمن بیماری های ریه آمریکا همراه با انجمن بیماری های ریه اروپا و انجمن جهانی سارکوئیدوز تعریف توصیفی زیر را پیشنهاد کرده اند. سارکوئیدوز یک اختلال چند ارگانی سیستم به علت (یا علل) ناشناخته است. معمولا جوانان یا بالغین میان سال را مبتلا می کند و غالبا با لنفادنوپاتی در ناف ریتین و انفیلتراسیون در ریه و ضایعات پوست و چشم تظاهر می کند. کبد، طحال، غدد لنفی ،غدد بزاق، قلب، سیستم عصبی، عضلات، استخوان ها و سایر اعضاء نیز ممکن است مبتلا شوند.آغاز سارکوئیدوز ممکن است به صورت حاد و آشکار و یا نهفته و با تظاهرات خفیف همراه باشد. شکل حاد با علایم عمومی و موضعی شدید بروز میکند و سیر آن معمولا کوتاهتر(چند ماه یا حدود یک سال)است و خود به خود با درمان به طرف بهبود و ضایعات و یا به طرف ازمان تثبیت میرود.یافته های بالینی دیگر شامل کاهش حجم اجبار بازدمی ،ظرفیت حیاتی، و ظرفیت انتظار هستند. آنمی، لنفوسیتوپنی ،بالارفتن میزان رسوب اریتروسیت ،افزایش تست های عملکرد کبدی ،هایپرکلسمی، و نفروپاتی هایپرکلسمیک نیز از یافته های معمول هستند. غدد لنفاوی آنها ممکن است متورم شده و شما متوجه توده هایی در زیر بازو، در گردن و یا در کشاله ران شوید. پزشک ممکن است متوجه تورم غدد لنفاوی در ناحیه قفسه سینه در زمان مشاهده عکس قفسه سینه شما بشود .پوست: شما ممکن است توده های کوچکی را درست درزیر پوستتان حبس کنید. همچنین ممکن است یک راش (لکه)ارغوانی رنگ و برجسته بر روی بینی، گونه ها، چانه و گوشها ایجاد شود. به این لکه ،lupus pernio گفته می شود.اریتم نودوزا نیز حالتی است که باعث ایجاد توده های قرمز و گردی اغلب در ساق پا می شود. این مساله ممکن است در واقع شروع بیماری سارکوئیدوز باشد و شایعترین نوع ضایعات پوستی در سارکوئیدوز می باشد.چشمها: سارکوئیدوز می تواند باعث التهاب چشمها و در نتیجه تاثیر بر روی دید شما شود. چشمها معمولا قرمز و دردناک میشوند. اگر شما مبتلا به سارکوئیدوز هستید و علایم چشمی در شما بروز کرد، حتما به پزشک مراجعه کنیدقلب: سارکوئیدوز باعث کند شدن کار قلب و یا منظم شدن ان می شود. آسیب به ریه ها که به علت سارکوئیدوز ایجاد شده می تواند تغییراتی در سمت راست قلب ایجاد کرده و آن را بزرگ نماید. این مساله می تواند در صورت عدم درمان باعث نارسایی سمت چپ قلب نیز شود. معمولا سارکوئیدوز باعث بزرگی قلب میشود که به آن کاردیومیوپاتی می گویند. این مساله به این معناست که قلب شما نمی تواند مثل قبل، قوی و موثر کار کند و به همین علت شما دچار تنگی نفس می شوید .اعصاب: سیستم عصبی شما ممکن است به چندین روش در سارکوئیدوز درگیر میشود. این بیماری می تواند باعث بروز سردرد ،مشکل در بلع، افتادگی صورت، نوعی مننژیت و مشکلات بینایی و شنوایی گردد. همچنین ممکن است بی حسی، احساس مورمور کردن و خواب رفتگی در صورت، دستها و پاهای شما ایجاد شود. در موارد نادر حتی ممکن است تشنج و یا سکته نیز بروز کند.کلیه ها : سارکوئیدوز تاثیر زیادی بر روی کلیه ها دارد .انسداد مجاری اشک و ضایعات آن باعث اشک ریزش می شود. بیمار ممکن است از آنوسمی شکایت داشته باشد.عوارض ناشی از این بیماری شاملعفونت قارچی ریهآب سیاه و کوری چشم )به ندرت(سنگ کلیه به علت افزایش کلسیم خون و یا ادرارپوکی استخوان و دیگر عوارض ناشی از مصرف کورتیکواستروئیدها برای مدت زمان طولانیپرفشاری خون ریویناباروری در مردان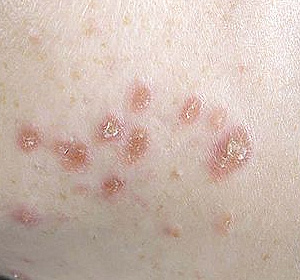 علت بیماری
در حالی که علت سارکوئیدوز مشخص نیست، عوامل ژنتیکی، عفونی و محیطی ممکن است در بروز بیماری دخالت داشته باشند. ویروس اپشتن بار ،کوکساکیB، پروپیونه باکتریوم آکنه، گونه های مایکوباکتریوم، آلومینیوم، زیرکونیوم، خاک رس و گرده درخت کاج همگی مثال هایی از عوامل مسبب این بیماری هستند. روش تشخیص بیماری چون علت سارکوئیدوز نامشخص است لذا بیماری وقتی که یافته های بالینی و رادیولوژیک منطبق با آن همراه با مدارک بافت شناسی که نشانه گرانولوم غیر کازفیه در بیشتر از یک ارگان وجود داشته باشد به صورت مطمئنی تائید میگردد مشروط به اینکه علل شناخته شده دیگری که سبب بیماری گرانولوماتوز میگردند یافت نگردد. تشخیص با مشاهدۀ رادیوگرافی غیرطبیعی قفسۀ صدری و یا افزایش آنزیم مبدل آنژیوتانسین ،لیزوزیم یا کلسیم سرم تائید م یشود. چهار هدف بررسی تشخیصی برای بیماران سارکوئیدوز عبارتند از 1)تایید هیستولوژیکی بیماری ،2)ارزیابی وسعت و شدت درگیری عضو ،3) ارزیابی این مسئله که آیا بیماری ثابت است یا احتمال پیشرفت دارد ،4) تشخیص این که درمان برای بیمار مفید خواهد بود یا نه. مطالعات ابتدایی شامل سابقه و آزمایش فیزیکی، آزمونهای عملکرد ریوی، شمارش کامل سلولهای خونی، خصوصیات شیمیایی سرم، و تست پوستی توبرکولین هستند. برای اثبات تشخیص سارکوئیدوز باید یافتههای بالینی و رادیولوژیک با مدرک بافتی یعنی گرانولم های متشکل از سلول های اپیتلیوئید بدون کازئوم همراه باشند. علایم بالینی سارکوئیدوز ممکن است بسته به نژاد بیمار و مزمن بودن بیماری، محل، وسعت درگیری عضو، و فعالیت گرانولوم به شی وههای مختلفی بروز کند.علایم غیر اختصاصی از قبیل تب، خستگی ،ناخوشی و کاهش وزن ممکن است در یک سوم بیماران مبتلا به سارکوئیدوز رخ دهد .درمانبرای بسیاری از بیماران، درمان سیستمیک ضروری نیست و تصمیم برای درمان سیستمیک بین مراکز درمانی مختلف است ،کورتیکواستروئیدها تکیه گاه اصلی درمان هستند اما دوز مناسب آن ها مشخص نیست .کورتیکو استروئیدها علایم را تخفیف میدهند و از تشکیل گرانولوم ها جلوگیری و میزان آنزیم آنژیوتانسین کانورتینگ را به حال طبیعی درمی آورند .کورتیکواستروئیدها درمان اصلی بیماری بوده و در افرادی که تهدید به نارسائی عضوی هستند یا بیماری در حال پیشرفت دارند مورد استفاده قرار میگیرد .درمان با کورتیکواستروئیدها ممکن است در ابتدا مؤثر باشد، اما عود شایع است. درمان طولانی مدت، استفاده از متوترکسات یا آزاتیوپرین را ایجاب می کند .درمان با کورتیکواستروئیدها ممکن است در ابتدا مؤثر باشد، اما عود شایع است. درمان طولانی مدت، استفاده از داروهای دیگری از جمله متوترکسات یا آزاتیوپرین را ایجاب می کند.پیش آگهی سیر و پیش آگهی این بیماری با چگونگی آغاز و وسعت آن مطابقت دارد. آغاز حاد با اریتماندوزوم یا لنفادنوپاتی ناف ریتین بدون علامت، حاکی از محدود شدن خود به خود آن است اما چنانچه آغاز آن نهفته باشد و به ویژه اگر چند عضو غیر از ریه مبتلا شوند به فیبروز ریه و فیبروز اعضا منجر خواهد شد. سیر بالینی بسیار متغیر بوده و میزان مرگ و میر از 1تا 5 درصد می باشد .مراقبت های لازم در معرض گرد و غبار و مواد شیمیایی که ممکن است به ریه ها آسیب برسانند، قرار نگیرید.رژیم متعادل داشته باشید. به مقدار زیاد میوه جات و سبزیجات و غلات را مصرف کنید.روزانه8 تا 10لیوان آب مصرف کنید6 تا 8ساعت در شب بخوابید. مرتب ورزش کنید.وزن خود را متعادل کنید.مصرف زنجبیل و روزماری مفید است.برای کاهش استرس می توانید از ویتامی نهای گروه B استفاده کنید.ماساژ گرمایی نیز برای این بیماران خوب است.ماساژ با روغن های تسکین دهنده مثل روغن روزماری ،کاج و یا اکالیپتوسبیماران مبتلا به سارکوئیدوز نباید در معرض گرد و غبار و مواد شیمیایی که ممکن است به ریه ها آسیب برسانند، قرار بگیرند .
علت بیماری
در حالی که علت سارکوئیدوز مشخص نیست، عوامل ژنتیکی، عفونی و محیطی ممکن است در بروز بیماری دخالت داشته باشند. ویروس اپشتن بار ،کوکساکیB، پروپیونه باکتریوم آکنه، گونه های مایکوباکتریوم، آلومینیوم، زیرکونیوم، خاک رس و گرده درخت کاج همگی مثال هایی از عوامل مسبب این بیماری هستند. روش تشخیص بیماری چون علت سارکوئیدوز نامشخص است لذا بیماری وقتی که یافته های بالینی و رادیولوژیک منطبق با آن همراه با مدارک بافت شناسی که نشانه گرانولوم غیر کازفیه در بیشتر از یک ارگان وجود داشته باشد به صورت مطمئنی تائید میگردد مشروط به اینکه علل شناخته شده دیگری که سبب بیماری گرانولوماتوز میگردند یافت نگردد. تشخیص با مشاهدۀ رادیوگرافی غیرطبیعی قفسۀ صدری و یا افزایش آنزیم مبدل آنژیوتانسین ،لیزوزیم یا کلسیم سرم تائید م یشود. چهار هدف بررسی تشخیصی برای بیماران سارکوئیدوز عبارتند از 1)تایید هیستولوژیکی بیماری ،2)ارزیابی وسعت و شدت درگیری عضو ،3) ارزیابی این مسئله که آیا بیماری ثابت است یا احتمال پیشرفت دارد ،4) تشخیص این که درمان برای بیمار مفید خواهد بود یا نه. مطالعات ابتدایی شامل سابقه و آزمایش فیزیکی، آزمونهای عملکرد ریوی، شمارش کامل سلولهای خونی، خصوصیات شیمیایی سرم، و تست پوستی توبرکولین هستند. برای اثبات تشخیص سارکوئیدوز باید یافتههای بالینی و رادیولوژیک با مدرک بافتی یعنی گرانولم های متشکل از سلول های اپیتلیوئید بدون کازئوم همراه باشند. علایم بالینی سارکوئیدوز ممکن است بسته به نژاد بیمار و مزمن بودن بیماری، محل، وسعت درگیری عضو، و فعالیت گرانولوم به شی وههای مختلفی بروز کند.علایم غیر اختصاصی از قبیل تب، خستگی ،ناخوشی و کاهش وزن ممکن است در یک سوم بیماران مبتلا به سارکوئیدوز رخ دهد .درمانبرای بسیاری از بیماران، درمان سیستمیک ضروری نیست و تصمیم برای درمان سیستمیک بین مراکز درمانی مختلف است ،کورتیکواستروئیدها تکیه گاه اصلی درمان هستند اما دوز مناسب آن ها مشخص نیست .کورتیکو استروئیدها علایم را تخفیف میدهند و از تشکیل گرانولوم ها جلوگیری و میزان آنزیم آنژیوتانسین کانورتینگ را به حال طبیعی درمی آورند .کورتیکواستروئیدها درمان اصلی بیماری بوده و در افرادی که تهدید به نارسائی عضوی هستند یا بیماری در حال پیشرفت دارند مورد استفاده قرار میگیرد .درمان با کورتیکواستروئیدها ممکن است در ابتدا مؤثر باشد، اما عود شایع است. درمان طولانی مدت، استفاده از متوترکسات یا آزاتیوپرین را ایجاب می کند .درمان با کورتیکواستروئیدها ممکن است در ابتدا مؤثر باشد، اما عود شایع است. درمان طولانی مدت، استفاده از داروهای دیگری از جمله متوترکسات یا آزاتیوپرین را ایجاب می کند.پیش آگهی سیر و پیش آگهی این بیماری با چگونگی آغاز و وسعت آن مطابقت دارد. آغاز حاد با اریتماندوزوم یا لنفادنوپاتی ناف ریتین بدون علامت، حاکی از محدود شدن خود به خود آن است اما چنانچه آغاز آن نهفته باشد و به ویژه اگر چند عضو غیر از ریه مبتلا شوند به فیبروز ریه و فیبروز اعضا منجر خواهد شد. سیر بالینی بسیار متغیر بوده و میزان مرگ و میر از 1تا 5 درصد می باشد .مراقبت های لازم در معرض گرد و غبار و مواد شیمیایی که ممکن است به ریه ها آسیب برسانند، قرار نگیرید.رژیم متعادل داشته باشید. به مقدار زیاد میوه جات و سبزیجات و غلات را مصرف کنید.روزانه8 تا 10لیوان آب مصرف کنید6 تا 8ساعت در شب بخوابید. مرتب ورزش کنید.وزن خود را متعادل کنید.مصرف زنجبیل و روزماری مفید است.برای کاهش استرس می توانید از ویتامی نهای گروه B استفاده کنید.ماساژ گرمایی نیز برای این بیماران خوب است.ماساژ با روغن های تسکین دهنده مثل روغن روزماری ،کاج و یا اکالیپتوسبیماران مبتلا به سارکوئیدوز نباید در معرض گرد و غبار و مواد شیمیایی که ممکن است به ریه ها آسیب برسانند، قرار بگیرند . 
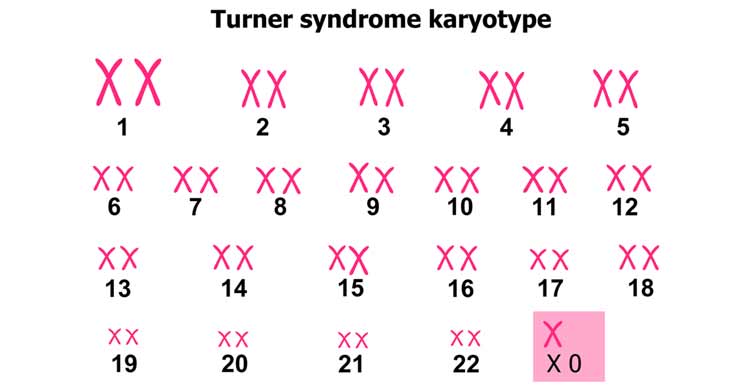 شیوع و همه گیری شناسی
این بیماری در ایالات متحده 1 مورد در هر 2000-2500 تولد مشاهده می شود یعنی حدود 800 مورد جدید تشخیصی در سال. در ایلات متحده در 75-80% موارد تک کروموزوم X از تخم مادر به جنین منتقل و اسپرم پدر که با تخم لقاح می یابد فاقد کروموزوم جنسی است.صرفا تعداد کمی بارداری در بین آن دسته از مبتلایان به TS که بطور خودبخود خونریزی قاعدگی داشته اند، گزارش شده است .3/1 فرزندان حاصله، دچار ناهنجاریهای مادرزادی مانند بیماری مادرزادی قلب، سندرم داون و ستون فقرات دو شاخه بوده اند .علت بیماری سندرم ترنر، اختلالی است که در تمام نژادها دیده می شود و ناشی از فقدان کامل یا نسبی کروموزوم x دوم در خانمهاست مونوزومی برای کروموزوم x بر اثر ناتوانی در گنجاندن یک کروموزوم x در یکی از گامتها یا از دست رفتن یک کروموزوم جنسی از گامت یا مراحل اولیه رویانی ایجاد میشود .علت بروز سندرم ترنر، رخداد “عدم انفصال” است. در طی تقسیم میتوز در هر والد رویداد عدم انفصال کروموزوم رخ داده که طی آن گامت یا اووسیت یا اسپرماتوسیت نه کروموزوم ایکس و نه کروموزوم y دارند. حال اگر این گامت ها با گامت والد دیگر لقاح یابد (کروموزوم نرمال دارد). رویان تشکیل شده دارای 45کروموزوم بوده و مونوزومی رخ میدهد.علایم ناشی از بیماری این افراد ظاهر کاملا زنانه دارند و با عدم وجود تخمدان( gonadal dysgenesi )وقد کوتاه مشخص می شوند. بیماران اغلب به سایر ناهنجاریهای مادرزادی مانند مشکلات آئورت، انسداد شریان ریوی، ناهنجاری های کلیوی، عقب ماندگی ذهنی و مشکلات شنوایی مبتلا هستند. سایر علایم بیماری عبارتند از :وجود بخشی پرده مانند در قسمت پوست بین گردن و شانهخط رویش موی پایین در پشت سرشکل غیر عادی چشمها از جمله افتادگی پلکادِِم لنفاوی اندام هانا هنجاری های اسکلتیسینه پهن و فاصله بیش از حد نوک پستان ها.تمام بیماران مبتلا به TS، کوتاه قد می باشند و بیش از 90 درصد آنها دیس ژنی تخمدان دارند. افراد دچار اختلال هوشی، معمولا واجد نوعی اختلال ساختمانی کروموزوم x هستند. از نظر اجتماعی، مبتلایان به TS معمولا خجالتی و منزوی هستند.روش تشخیص در نیمی از موارد تشخیص در ماههای اول زندگی به وسیله مشخصات فیزیکی صورت میگیرد. در سایر بیماران در دوران نوجوانی به علت رشد پایین تر از حد نرمال یا عدم رسیدن به بلوغ صورت میگیرد.زمانی که پزشک به سندرم ترنر مشکوک میشود میتواند با انجام کاریو تایپ (آنالیز کروموزوم) تشخیص را به طورقطعی تایید کند. سندرم ترنر می تواند در طول حاملگی به وسیله CVS یا آمینیو سنتز (نمونه گیری از مایع رحمی) مشخص شود.تشخیص این سندرم قبل از تولد جنین به واسطه آمینوسنتز امکان پذیر است و حتی ناهنجاریهای قلبی و کلیوی نیز با انجام سونوگرافی مشهود میگردد. اما این سندرم معمولا در ابتدای تولد و یا هنگامیکه انتظار میرود که بلوغ رخ داده باشد، تشخیص داده میشود .اگر کودک دارای چندین نشانه از سندرم ترنر باشد، آزمایش خون صورت گرفته و کاریوتیپ صورت می گیرد. این تست تعداد کروموزومهای فرد را شمارش کرده و هر گونه تغییر شکل ناهنجار و یا فقدان هر بخش از کروموزوم را تشخیص میدهد. در برخی موارد هیچ گونه علائم بالینی در ابتدای تولد مشهود نمی باشد و این امر تا رسیدن به زمان بلوغ دختر به تعویق می افتد. با این حال مهم است که این سندرم زودتر تشخیص داده شود تا هر چه زودتر درمانهای هورمونی لازم صورت گیرد. تشخیص زودرس و درمان به پیشگیری از سایر مشکلات بخصوص عوارض مزمن از قبیل استئوپورز و دیابت کمک خواهد کرد. استفاده متناوب از اولتراسوند می تواند در تعیین بیماری به وسیله نشانه های فیزیکی قبل از تولد کمک نماید .درمان تزریق هورمون رشد اگر به موقع انجام شود، می تواند با افزایش رشد، حداکثر قد در دوران بلوغ را افزایش دهد. درمان با استروژن معمولاً در سنین 12 یا 13 سال آغاز می شود و می تواند در بروز خصوصیات ثانویه جنسی موثر باشد اما نمی تواند موجب درمان نازایی شود. امروزه به زنان مبتلا به TS کمک می کنند که باردار شوند. به این ترتیب که یک تخمک اهدایی بارور شده را پرورش داده و رویان را در رحم بیمار ترنری جاسازی می کنند. به کمک هورمون درمانی، این زن می تواند جنین آزمایشگاهی را حفظ نموده و زایمان کند.مراقبت های لازم بسیار ضروری است که دختر خود را برای معالجه به نزد پزشک خانوادگی و یا متخصص اطفال برده و او را تحت معاینه و مداوا قرار دهید. زیرا که پزشک او در طول زندگی بیمار هماهنگی لازم برای انجام سایر خدمات را در میان تعدادی از متخصصان به وجود می آورد.معاینات منظم پیشرفت های قابل توجهی در کیفیت و طول عمر زنان مبتلا به سندرم را به وجود آورده است.عادات زندگی سالم، حفظ وزن مناسب و ورزش منظم در طول زندگی مهم هستند. در طول این دوره مشاوره با متخصص قلب و حصول اطمینان از سلامت بافت قلبی و فعالیت آن اهمیت دارد. پیش آگهی این ناهنجاری کروموزومی با مشکلات و عوارض بسیاری همراه است علاوه بر فقدان بلوغ جنسی اگر نقص قلبی شدیدی برای نوزاد مبتلا به سندرم ترنر حادث نشده باشد بسیاری از دختران مبتلا تا دوران بزرگسالی زنده می مانند. در حدود 2 تا 5 درصد زنان مبتلا به این سندرم، دارای مشکلات تخمدانی هستند. مبتلایان به این سندرم ممکن است در آینده از عوارضی مانند ناهنجاری های کلیه، فشار خون بالا، چاقی، دیابت، کاتاراکت، آرتریت و اسکلیوزیس رنج ببرند.
شیوع و همه گیری شناسی
این بیماری در ایالات متحده 1 مورد در هر 2000-2500 تولد مشاهده می شود یعنی حدود 800 مورد جدید تشخیصی در سال. در ایلات متحده در 75-80% موارد تک کروموزوم X از تخم مادر به جنین منتقل و اسپرم پدر که با تخم لقاح می یابد فاقد کروموزوم جنسی است.صرفا تعداد کمی بارداری در بین آن دسته از مبتلایان به TS که بطور خودبخود خونریزی قاعدگی داشته اند، گزارش شده است .3/1 فرزندان حاصله، دچار ناهنجاریهای مادرزادی مانند بیماری مادرزادی قلب، سندرم داون و ستون فقرات دو شاخه بوده اند .علت بیماری سندرم ترنر، اختلالی است که در تمام نژادها دیده می شود و ناشی از فقدان کامل یا نسبی کروموزوم x دوم در خانمهاست مونوزومی برای کروموزوم x بر اثر ناتوانی در گنجاندن یک کروموزوم x در یکی از گامتها یا از دست رفتن یک کروموزوم جنسی از گامت یا مراحل اولیه رویانی ایجاد میشود .علت بروز سندرم ترنر، رخداد “عدم انفصال” است. در طی تقسیم میتوز در هر والد رویداد عدم انفصال کروموزوم رخ داده که طی آن گامت یا اووسیت یا اسپرماتوسیت نه کروموزوم ایکس و نه کروموزوم y دارند. حال اگر این گامت ها با گامت والد دیگر لقاح یابد (کروموزوم نرمال دارد). رویان تشکیل شده دارای 45کروموزوم بوده و مونوزومی رخ میدهد.علایم ناشی از بیماری این افراد ظاهر کاملا زنانه دارند و با عدم وجود تخمدان( gonadal dysgenesi )وقد کوتاه مشخص می شوند. بیماران اغلب به سایر ناهنجاریهای مادرزادی مانند مشکلات آئورت، انسداد شریان ریوی، ناهنجاری های کلیوی، عقب ماندگی ذهنی و مشکلات شنوایی مبتلا هستند. سایر علایم بیماری عبارتند از :وجود بخشی پرده مانند در قسمت پوست بین گردن و شانهخط رویش موی پایین در پشت سرشکل غیر عادی چشمها از جمله افتادگی پلکادِِم لنفاوی اندام هانا هنجاری های اسکلتیسینه پهن و فاصله بیش از حد نوک پستان ها.تمام بیماران مبتلا به TS، کوتاه قد می باشند و بیش از 90 درصد آنها دیس ژنی تخمدان دارند. افراد دچار اختلال هوشی، معمولا واجد نوعی اختلال ساختمانی کروموزوم x هستند. از نظر اجتماعی، مبتلایان به TS معمولا خجالتی و منزوی هستند.روش تشخیص در نیمی از موارد تشخیص در ماههای اول زندگی به وسیله مشخصات فیزیکی صورت میگیرد. در سایر بیماران در دوران نوجوانی به علت رشد پایین تر از حد نرمال یا عدم رسیدن به بلوغ صورت میگیرد.زمانی که پزشک به سندرم ترنر مشکوک میشود میتواند با انجام کاریو تایپ (آنالیز کروموزوم) تشخیص را به طورقطعی تایید کند. سندرم ترنر می تواند در طول حاملگی به وسیله CVS یا آمینیو سنتز (نمونه گیری از مایع رحمی) مشخص شود.تشخیص این سندرم قبل از تولد جنین به واسطه آمینوسنتز امکان پذیر است و حتی ناهنجاریهای قلبی و کلیوی نیز با انجام سونوگرافی مشهود میگردد. اما این سندرم معمولا در ابتدای تولد و یا هنگامیکه انتظار میرود که بلوغ رخ داده باشد، تشخیص داده میشود .اگر کودک دارای چندین نشانه از سندرم ترنر باشد، آزمایش خون صورت گرفته و کاریوتیپ صورت می گیرد. این تست تعداد کروموزومهای فرد را شمارش کرده و هر گونه تغییر شکل ناهنجار و یا فقدان هر بخش از کروموزوم را تشخیص میدهد. در برخی موارد هیچ گونه علائم بالینی در ابتدای تولد مشهود نمی باشد و این امر تا رسیدن به زمان بلوغ دختر به تعویق می افتد. با این حال مهم است که این سندرم زودتر تشخیص داده شود تا هر چه زودتر درمانهای هورمونی لازم صورت گیرد. تشخیص زودرس و درمان به پیشگیری از سایر مشکلات بخصوص عوارض مزمن از قبیل استئوپورز و دیابت کمک خواهد کرد. استفاده متناوب از اولتراسوند می تواند در تعیین بیماری به وسیله نشانه های فیزیکی قبل از تولد کمک نماید .درمان تزریق هورمون رشد اگر به موقع انجام شود، می تواند با افزایش رشد، حداکثر قد در دوران بلوغ را افزایش دهد. درمان با استروژن معمولاً در سنین 12 یا 13 سال آغاز می شود و می تواند در بروز خصوصیات ثانویه جنسی موثر باشد اما نمی تواند موجب درمان نازایی شود. امروزه به زنان مبتلا به TS کمک می کنند که باردار شوند. به این ترتیب که یک تخمک اهدایی بارور شده را پرورش داده و رویان را در رحم بیمار ترنری جاسازی می کنند. به کمک هورمون درمانی، این زن می تواند جنین آزمایشگاهی را حفظ نموده و زایمان کند.مراقبت های لازم بسیار ضروری است که دختر خود را برای معالجه به نزد پزشک خانوادگی و یا متخصص اطفال برده و او را تحت معاینه و مداوا قرار دهید. زیرا که پزشک او در طول زندگی بیمار هماهنگی لازم برای انجام سایر خدمات را در میان تعدادی از متخصصان به وجود می آورد.معاینات منظم پیشرفت های قابل توجهی در کیفیت و طول عمر زنان مبتلا به سندرم را به وجود آورده است.عادات زندگی سالم، حفظ وزن مناسب و ورزش منظم در طول زندگی مهم هستند. در طول این دوره مشاوره با متخصص قلب و حصول اطمینان از سلامت بافت قلبی و فعالیت آن اهمیت دارد. پیش آگهی این ناهنجاری کروموزومی با مشکلات و عوارض بسیاری همراه است علاوه بر فقدان بلوغ جنسی اگر نقص قلبی شدیدی برای نوزاد مبتلا به سندرم ترنر حادث نشده باشد بسیاری از دختران مبتلا تا دوران بزرگسالی زنده می مانند. در حدود 2 تا 5 درصد زنان مبتلا به این سندرم، دارای مشکلات تخمدانی هستند. مبتلایان به این سندرم ممکن است در آینده از عوارضی مانند ناهنجاری های کلیه، فشار خون بالا، چاقی، دیابت، کاتاراکت، آرتریت و اسکلیوزیس رنج ببرند. 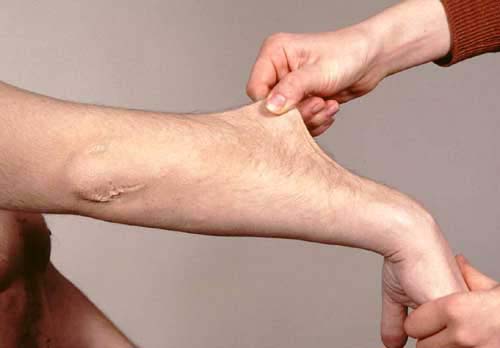
 میزان شیوع این بیماری از 1 در250000تا 1 در 50000در جمعیت عمومی برآورد شده است. اما مطالعات صورت گرفته در مناطق وسیع تر حاکی از افزایش آمار این بیماری است و محققان معتقدند گستره این بیماری وسیع تر از آمار برآورد شده می باشد چرا که تنوع این بیماری بیشتر است. سندرم اهلر دانلوس در تمام قومها و نژادها بین زنان و مردان به نسبت مساوی شایع است .هر چند بعضی از انواع آن در بین گروههای خاصی رایج ترند.علت بیماری انواع مختلفی از این سندرم بر پایه نوع تغییری است که در ژنهای فرد رخ داده است. مهمترین چالشی که در این سندرم وجود دارد ،پروتئین های فیبری هستند که باعث الاستیسیته و استحکام بافت همبند میگردد. این موتاسیون ژنتیکی باعث تغییر در عملکرد آنزیمی میگردد که فقدان آن باعث ضعف و ناپایداری بافت همبند میگردد. بیشترین فرم ابتلا به این سندرم در اثر حالتی رخ می دهد که به “الگوی اتوزومال غالب” معروف است. این بدان معناست که برای ابتلا به این سندرم تنها وجود یک کپی از این ژن ناقص کافی خواهد بود. در این صورت احتمال 50 درصد وجود دارد که کودکی به این سندرم مبتلا گردد و این ژن جهش یافته را به ارث ببرد .علایم ناشی از بیماری علائم و نشانه های رایج این سندرم عبارتند از:انعطاف زیاد و یا شکنندگی پوستروند غیر نرمال ترمیم زخم که بیش از حد معمول زمان لازم دارندانعطاف پذیری بیش از حد مفاصل که دامنه حرکتی بیش از حد نرمال خود دارند.در رفتگی، شانه، هیپ، زانو و انگشتانضعف ماهیچه ایتاخیر تکامل حرکتیکبود شدگی پوستمشکلات قلبی و گسیختگی خودبخودی دیواره عروقسابقه خانوادگی، آنوریسم مغزی و یا کولونروش تشخیص تشخیص این سندرم بر پایه، یافتههای بالینی بیماران و سابقه خانوادگی آنان می باشد. در برخی از انواع این سندرم بیوپسی پوست به منظور بررسی ترکیبات شیمیایی بافتهای همبند مورد استفاده قرار میگیرد.تستهای ژنتیکی: تست DNA برای انواع کلاسیک، عروقی، کیفواسکلیوزیس، مورد استفاده قرار میگیرد.تست ادراری: این تست برای تشخیص فرم کیفواسکلیوز موثر خواهد بود. این تست میزان آنزیمی که به واسطه بروز این فرم از سندرم تولید می شود را اندازه گیری میکند.بیوپسی پوست: در این تست مقداری نمونه پوستی گرفته و در زیر میکروسکپ مورد بررسی قرار می گیرد. این تست به تشخیص ناهنجاریهایی در ساختار فیبری پوست مفید خواهد بود .اولتراسوند قلبی: بررسی به منظور تشخیص پرولاپس میترال
میزان شیوع این بیماری از 1 در250000تا 1 در 50000در جمعیت عمومی برآورد شده است. اما مطالعات صورت گرفته در مناطق وسیع تر حاکی از افزایش آمار این بیماری است و محققان معتقدند گستره این بیماری وسیع تر از آمار برآورد شده می باشد چرا که تنوع این بیماری بیشتر است. سندرم اهلر دانلوس در تمام قومها و نژادها بین زنان و مردان به نسبت مساوی شایع است .هر چند بعضی از انواع آن در بین گروههای خاصی رایج ترند.علت بیماری انواع مختلفی از این سندرم بر پایه نوع تغییری است که در ژنهای فرد رخ داده است. مهمترین چالشی که در این سندرم وجود دارد ،پروتئین های فیبری هستند که باعث الاستیسیته و استحکام بافت همبند میگردد. این موتاسیون ژنتیکی باعث تغییر در عملکرد آنزیمی میگردد که فقدان آن باعث ضعف و ناپایداری بافت همبند میگردد. بیشترین فرم ابتلا به این سندرم در اثر حالتی رخ می دهد که به “الگوی اتوزومال غالب” معروف است. این بدان معناست که برای ابتلا به این سندرم تنها وجود یک کپی از این ژن ناقص کافی خواهد بود. در این صورت احتمال 50 درصد وجود دارد که کودکی به این سندرم مبتلا گردد و این ژن جهش یافته را به ارث ببرد .علایم ناشی از بیماری علائم و نشانه های رایج این سندرم عبارتند از:انعطاف زیاد و یا شکنندگی پوستروند غیر نرمال ترمیم زخم که بیش از حد معمول زمان لازم دارندانعطاف پذیری بیش از حد مفاصل که دامنه حرکتی بیش از حد نرمال خود دارند.در رفتگی، شانه، هیپ، زانو و انگشتانضعف ماهیچه ایتاخیر تکامل حرکتیکبود شدگی پوستمشکلات قلبی و گسیختگی خودبخودی دیواره عروقسابقه خانوادگی، آنوریسم مغزی و یا کولونروش تشخیص تشخیص این سندرم بر پایه، یافتههای بالینی بیماران و سابقه خانوادگی آنان می باشد. در برخی از انواع این سندرم بیوپسی پوست به منظور بررسی ترکیبات شیمیایی بافتهای همبند مورد استفاده قرار میگیرد.تستهای ژنتیکی: تست DNA برای انواع کلاسیک، عروقی، کیفواسکلیوزیس، مورد استفاده قرار میگیرد.تست ادراری: این تست برای تشخیص فرم کیفواسکلیوز موثر خواهد بود. این تست میزان آنزیمی که به واسطه بروز این فرم از سندرم تولید می شود را اندازه گیری میکند.بیوپسی پوست: در این تست مقداری نمونه پوستی گرفته و در زیر میکروسکپ مورد بررسی قرار می گیرد. این تست به تشخیص ناهنجاریهایی در ساختار فیبری پوست مفید خواهد بود .اولتراسوند قلبی: بررسی به منظور تشخیص پرولاپس میترال